

Epilepsy Insights

Gene therapy for Dravet syndrome – 2020 update
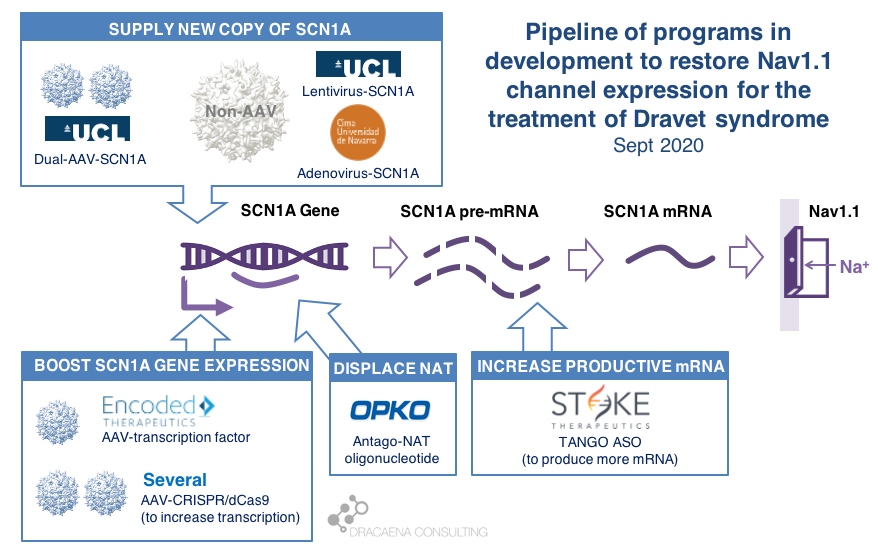
There are multiple gene therapy and oligonucleotide programs in development for Dravet syndrome including those that supply and extra copy of the SCN1A gene and those that boost expression from the healthy SCN1A gene copy. Clinical trials have already started, with Stoke Therapeutics initiating the first clinical trial with a disease-targeting therapy in Dravet syndrome in summer 2020. Behind Stoke, gene therapies are approaching the clinic with Encoded Therapeutics having the most advanced clinical candidate and preparing for trials in 2021.
In February of 2019 I reviewed the state of development of gene therapy approaches for Dravet syndrome at that time. A lot has changed in a year a half, for good. So I felt this update was long due.
Here is a review of the gene therapies in development for treating Dravet syndrome, how each of them works, and the timings that we anticipate for clinical trials.
CURRENT GENE THERAPIES IN DEVELOPMENT FOR DRAVET SYNDROME
In diseases like Dravet syndrome where the problem is that a copy of the gene is missing or not functional due to mutations, the desired therapy is one that can restore normal gene expression and therefore normal protein production. In other words, we need more protein.
In the case of Dravet syndrome, the gene is SCN1A, and the protein that is needed is the neuronal sodium channel Nav1.1. As a result of mutations in the gene, the number of Nav1.1 channels at the neuronal surface is not sufficient, there is less sodium crossing the membrane, and the neuron cannot fire properly. The result is Dravet syndrome.
One particularity of Dravet syndrome is that only one of the two copies of the SCN1A gene is affected, the second one is perfectly fine, so that second copy can serve as the supply for extra protein production. As you will see, the most advanced programs are exploiting this possibility.
Broadly speaking, there are two potential approaches to restore protein expression in Dravet syndrome: you either supply the cell with an extra healthy copy the gene, which will lead to more protein being produced, or you try to boost the expression from the healthy gene.
(1) Supply a new copy of SCN1A
When people think about “gene therapy”, the type of therapy they are thinking about is the one where the DNA of a virus gets replaced by the gene that the person needs, and that modified virus is used as a Trojan horse to infect cells and deliver them the therapeutic gene.
The most commonly used virus for gene therapy is the Adeno-Associated Virus (AAV), and because we have so much experience with gene therapies being developed and approved using AAV, this virus is the first choice for most in the gene therapy space. But AAVs are small virus, and have a limited capacity for the size of genes that they can carry, and SCN1A happens to be way too large for using these virus to carry the gene. In January of 2018, before we knew about the new gene therapies currently in development for Dravet syndrome, I reviewed this problem in the article “big gene, small virus”.
One way around this is to use other viruses that are larger and can therefore carry larger genes inside. One of these is the Adenovirus, which we all hear much about recently because it is also the virus of choice for the AztraZeneca vaccine in development for COVID-19.
|| The Spain-France-Israel consortium CureDravet is developing a gene therapy for Dravet syndrome using Adenovirus, a type of high-capacity virus that is large enough to contain the entire SCN1A gene. The leading Spanish group from Rubén Hernández has presented early results at conferences (e.g. European Paediatric Neurology Society (EPNS) 2019) showing rescue of multiple disease phenotypes in transgenic mice by administration of an adenovirus-based SCN1A gene therapy. As of the last public update this is still a preclinical program working towards the development of a clinical candidate.
Another way around the gene-vs-virus size issue is to use not one, but two AAVs, and to make each carry half of the SCN1A gene. This is a strategy that is also being explored for gene editing using CRISPR because the CRISPR approach requires expressing in the tissue proteins whose genes are too large to fit into an AAV. So the strategy of two AAVs is being explored both for rare genetic diseases with large genes such as Dravet syndrome, and for gene editing in general.
|| At UCL, the team of Rajvinder Karda and Simon Waddington is working on two approaches. One is to use another type of large-capacity virus, Lentivirus, to carry the SCN1A gene. The second approach uses two AAV virus, each containing half of the SCN1A gene, which are able to recreate the full channel once they co-infect the same cells. As of the last public updates these are also still preclinical programs working towards the development of clinical candidates.
(2) Boost expression of SCN1A
Another strategy to restore Nav1.1 levels is to target the good SCN1A gene copy or some stem downstream of it to increase protein production, without needing to add an external gene copy with a virus. This one is the strategy most advanced for Dravet syndrome.
There are several ways to do this, and luckily for us many of these approaches are being pursued and some of them are already in clinical development or getting very close.
As a small biology primer: genes (like SCN1A) are large stretches of DNA that contain the information needed to produce proteins (like Nav1.1). These large stretches of DNA are first copied into large stretches of RNA (pre-messenger RNA or pre-mRNA), then some sections that are not meant to be part of the final protein are removed from the RNA to make it shorter (mRNA), and then the mRNA is read to produce the final Nav1.1 protein.
What happens in Dravet syndrome as a result of having a mutation (or deletion) in one copy of the SCN1A is the following:

By now we know that the levels of Nav1.1 protein production are regulated in the cell at different levels along this sequence of steps, offering scientists multiple points where to act when trying to boost production of Nav1.1:
Regulation at the step of gene expression 1: the SCN1A gene has sequences around it that tell the cell how much the gene should be copied and therefore how much RNA should be produced. These are promoter or enhancer or other regulatory regions that could be targeted to boost gene expression and protein production.
Regulation at the step of gene expression 2: cells produce some RNAs that are not meant to produce proteins, but to match the sequence of some genes and prevent them from being read by the cell. SCN1A happens to be the target of one of those Natural Antisense Transcripts, or NAT. This endogenous “repressor” of SCN1A expression could be also targeted to boost gene expression and protein production.
Regulation at the step of RNA processing: neurons produce a large amount of pre-mRNA of SCN1A, with only a fraction of it being processed into mRNA which is the final form that will be used to produce proteins. This creates a “reserve pool” of pre-mRNA that could potentially also be targeted to get more productive mature mRNA and therefore boost protein production.

The first company to try one of these approaches was OPKO Health. OPKO decided to go after the Natural Antisense Transcripts that limits SCN1A expression by developing an oligonucleotide that could displace it, releasing the good SCN1A copy and leading to more mRNA and more Nav1.1 protein levels. Because their oligonucleotide antagonizes the NAT for SCN1A they called it an AntagoNAT (OPK88001, previously CUR-1916). While OPKO indicated plans to initiate clinical trials as early as in 2017, these timelines were moved to 2018 and then 2019 and as of summer 2020 there are no news of whether this program is still active. In the meantime, other corporate programs have appeared and progressed further into the clinic.
As a side note: oligonucleotide therapeutics are NOT gene therapies, if we stick to the actual meaning of gene therapies using genes as therapies. But because oligonucleotide therapeutics target gene expression or mRNA processing leading to more protein production we often bundle them together with the “real gene therapies”. Both are approaches that target the genetic problem in the disease, either correcting it or compensating for it (like when boosting the healthy gene copy expression). Both are expected to result in increased levels of Nav1.1. And both are expected to lead to transformational improvements across different symptom domains in patients. So for all purposes it makes sense to talk about together when reviewing gene therapies.
2018 brought the good news that Stoke Therapeutics was developing an antisense oligonucleotide treatment to boost expression of SCN1A as well. This oligonucleotide binds to the pre-mRNA, facilitating the transformation of some of the pre-mRNA “reserve pool” into mature mRNA and leading to more Nav1.1 protein. Stoke recently published their preclinical proof of concept (July 2020 and August 2020), showing how they were able to produce an antisense oligonucleotide able to trigger this pre-mRNA to mRNA processing specifically for SCN1A and not affecting other sequence-related sodium channel genes. Their therapeutic candidate, called STK-001, increased expression of the SCN1A mRNA and Nav1.1 protein in mice, and had remarkable efficacy in mice with Dravet syndrome caused by SCN1A haploinsufficiency.
|| Since my early 2019 update, Stoke Therapeutics had an IPO, obtained an FDA Orphan Drug Designation for STK-001, and in August of 2020 it initiated a Phase 1/2s study in patients 2 to 18 years with Dravet syndrome. The dosing of the first patient in this study represented a historical moment for the Dravet syndrome field: for the first time, a person born with Dravet syndrome was given a therapy designed to correct their genetic problem (SCN1A haploinsufficiency). That day the field moved from developing and testing symptomatic treatments to developing and testing disease-targeting treatments, which the patient community often prefers to call cures.
The program from Stoke Therapeutics is the most advanced of all gene therapies and gene therapy-like approaches in development for Dravet syndrome. The second program in line is a gene therapy approach by Encoded Therapeutics that was still in stealth mode in 2018.
|| In June 2019, Encoded Therapeutics emerged from stealth mode with a series C of $104M with a lead program for Dravet syndrome. Encoded managed to beat the gene-vs-virus size problem by using AAV, the gold-standard viral vector for gene therapy, to deliver to the brain a synthetic transcription factor for SCN1A. In other words, the SCN1A gene might be too large, but we can still AAV to deliver to the brain the gene of a specific SCN1A booster. In barely one year, Encoded has closed another funding round of $135M, obtained the Orphan Drug Designation and Rare Pediatric Disease Designation by the FDA for their ETX101 gene therapy for Dravet syndrome, and anticipates to start clinical trials in 2021.
Several academic efforts follow Stoke and Encoded in the pursue of boosting expression from the good SCN1A gene copy. All of these projects are in early preclinical stages, and they have not yet published a solid proof of concept in a Dravet syndrome mouse model, which is an initial stage prior to advancing the treatments towards clinical trials. These programs are therefore all years away from clinical trial initiation, with no guarantee of succeeding.
|| An academic group in Italy, with funding from CURE and the Dravet Syndrome European Federation, is researching an oligonucleotide approach to boost production of Nav1.1 protein though a combination of transcriptional and translational activity. There have been no recent public updates on the progress of this project.
|| The Vania Broccoli lab in Italy in collaboration with Gabriele Lignani lab at UCL are using a CRISPR approach to target the promoter of SCN1A and increase its activity, therefore boosting production of Nav1.1 from the good SCN1A gene copy. They use a variant of CRISPR that uses dead Cas9 to find – but not to cut – the desired genomic region and activate it. They have demonstrated the ability of upregulating Nav1.1 using this system using lentivirus in vitro, and published an early proof of principle in mice by using two AAVs to deliver all of the different elements of the CRISPR system into the brain, which would not otherwise fit into one AAV. It is early to know if these dual-AAV approaches will achieve sufficient biodistribution and expression in the brain, or if single-AAV or single-Adenovirus approaches are needed, probably reducing the size of the cargo like Encoded is doing with their transcription factor approach.
|| The Yamakawa lab in Japan has also published a recent study using CRISPR with dead Cas9 to increase SCN1A gene expression and protein production in mice. For multiple reasons, their approach cannot be considered a proof of concept for the future clinical treatment. For example they used a type of AAV (PHP.B), which has fantastic biodistribution in mice but not in primates, and administered it via intravenous administration. Peripheral administration of AAV to target neurological diseases also seems an approach less viable in patients than in mice. In this case part of the CRISPR system was genetically encoded in the mice, so they only needed administration of one AAV. This would also not be possible in patients, who would need the entire treatment to be delivered to them using suitable route of administration and virus type. But what this study provides is a proof of biologic rescue of SCN1A expression in GABAergic neurons starting at 4 weeks of age in mice, and the improvement in seizures, mortality and behavioral phenotypes that result from such rescue.
|| SCN1A encodes for the alpha subunit of Nav1.1, so less alpha subunit means less mature sodium channels, but there is also a beta subunit involved in channel formation which is encoded by SCN1B. The Hampson lab in Toronto recently published a gene therapy approach using AAV to deliver an additional copy of SCN1B, which is a small gene that fits the virus. The reasoning is that upregulating the beta subunit might be able to drag more alpha subunits to the surface and result in more total Nav1.1. In the study, funded by Dravet Canada and the US Dravet Syndrome Foundation, mice heterozygous for SCN1A had improvements in mortality, seizures and behavioral outcomes, although some of these were more pronounced in males or females. None of the SCN1A boosting approaches had reported any sex-bias when it comes to efficacy. The beta subunit produced by SCN1B is auxiliary to multiple channels, not unique to Nav1.1, and the study did not determine which of these channels might be mediating efficacy, so the relevance of this study to the SCN1A-targeted therapeutics is unclear.
FINAL THOUGHTS
There are several differences between oligonucleotide treatments and gene therapies. Some of these are differences that are more important for us scientists. For example to better understand the need to increase SCN1A expression in all cells that naturally express the gene (what oligonucleotides do) versus the convenience of boosting it only in those that are the most affected in the disease (an option available only for gene therapies because they can use cell-specific promoters). Other differences are more important to patient families, such as the need to repeat dosing with oligonucleotide treatments, usually spaced out by several months, versus the once-and-done single-administration approach of the gene therapies. In the end what we all want to see is which approach produces the most clinical improvement with the least side effects and burden of administration, and it is very likely that we will want to have multiple options in the market.
When thinking about these options, the field of SMA comes to mind. Spinraza (by Biogen) is an ASO able to increase the production of SMN, and a total game-changer in a terrible disease that used to kill babies. Then Zolgensma (by Novartis) reached the market, as the first gene therapy using AAV to deliver a copy of the SMN1 gene. With both in the market we have started seeing clinical studies to evaluate the combination of the ASO and the gene therapy in patients with SMA. And last month FDA approved risdiplam (Evrysdi) from Genentech/Roche which is a small molecule that can be taken orally and that increases the production of SMN. In 4 years the field of SMA has gone from facing a death sentence to having three very different modalities all able to rescue the missing protein expression (ensuring access to these treatments is a different topic, but I still want to highlight that there is a lot of work to do there).
So while we keep an eye on Stoke and Encoded and the development of their two very different approaches, we might want to keep another eye following the developing of small molecule activators of the Nav1.1 channel, for which there are several programs in the development.
We also want to make sure that the current oligonucleotide treatments and gene therapies in development are successful. A major challenge they face is that we don’t know how to design a clinical trial with a gene therapy in Dravet syndrome. So far clinical trials measuring seizure frequency have been very successful, but a gene therapy is expected to improve the syndrome beyond just seizure frequency. The field of Dravet syndrome is still immature when it comes to clinical outcome measure development and validation for non-seizure outcomes (for non-scientists in the audience: we don’t know how to quantify improvements of the disease in a clinical trial beyond seizures). Both Stoke and Encoded are running observational studies that will hopefully identify the best outcome measures and endpoints that will be needed for pivotal studies.
And another development that I miss is that despite so many approaches trying to increase the levels of Nav1.1 we don’t have any biomarker that could help us measure the levels of functional or total Nav1.1 in patients. This will make it hard to interpret clinical trials where a dose of the treatment is ineffective based on the selected outcome measures. And having a biomarker for protein levels will provide early data to encourage longer patient monitoring as we wait for some of those outcome measures to show enough of a change. So in parallel to a race for developing new treatments, we are seeing a race to de-risk the field fast enough so that those treatments get their best shot at succeeding in a pivotal trial.
IN SUMMARY
There are multiple gene therapy and oligonucleotide programs in development for Dravet syndrome including those that supply and extra copy of the SCN1A gene and those that boost expression from the healthy SCN1A gene copy.
Clinical trials have already started, with Stoke Therapeutics initiating the first clinical trial with a disease-targeting therapy in Dravet syndrome in summer 2020.
Behind Stoke, gene therapies are approaching the clinic with Encoded Therapeutics having the most advanced clinical candidate and preparing for trials in 2021.
I look forward to seeing precompetitive collaborations around the common challenges of validating clinical outcome measures and biomarkers, which are needed to maximize the success of gene therapies for Dravet syndrome.
Ana Mingorance PhD

Main lessons from the European Congress on Epilepsy 2018
Every other year the International League Against Epilepsy organises a major epilepsy medical congress in Europe called the European Congress on Epilepsy (ECE). This year I attended the main three days of the ECE addition in Vienna, looking at the field partly as a drug developer and partly as a patient advocate working on behalf of rare epilepsy patient communities. Here is the list of what I found the most interesting at the ECE 2018 meeting.
Every other year the International League Against Epilepsy organises a major epilepsy medical congress in Europe called the European Congress on Epilepsy (ECE). This year I attended the main three days of the ECE addition in Vienna, looking at the field partly as a drug developer and partly as a patient advocate working on behalf of rare epilepsy patient communities.
Here is the list of what I found the most interesting at the ECE 2018 meeting:
1. The big players are gone, the orphan players are taking over.
I missed seeing the large stands in the exhibition hall that UCB or GSK would always have. Instead we are now surrounded by (often) smaller companies that are launching molecules to treat orphan forms of epilepsy. It is an evolution that we also see in other disease fields, and that started becoming clear in epilepsy a couple of years ago. As rare diseases become more popular for drug development and we have more approvals, not only we see more orphan drugs at epilepsy conventions but we also see different players.
If you turned around at the ECE 2018 meeting you could see the stands of GW Pharmaceuticals, fresh from their FDA approval for Epidiolex for treating Dravet and Lennox-Gastaut syndrome (expected EMA decision 1H 2019); Biomarin, which develops Brineura, with a stand focused on CLN2 (one of Batten disease types); Biocodex, with the newly FDA-approved stiripentol for treating Dravet syndrome (approved in EU since 2007); and Novartis with a stand on Tuberous Sclerosis Complex for their also recent approval of Votubia/Afinitor.
In addition to those, Zogenix, in very late stages of development of fenfluramine for the treatment of Dravet syndrome, didn’t have a stand but sponsored a symposium on Dravet and had multiple posters. I would expect to see Zogenix and potentially Ovid Therapeutics get some more visibility at the next American Epilepsy Society meeting given their active late-stage programs in Dravet and Lennox-Gastaut syndromes. But for now, at the European meeting, we could already see four orphan epilepsy syndromes replacing much of the exhibition floor space formerly dedicated to medicines with a much broader spectrum. Since I focus on rare epilepsy syndromes, seeing that many drugs coming to the market and so much research on rare syndromes with epilepsy makes me very happy.

2. Epileptologists discussing much more than just epilepsy.
Probably as a combination of paying more attention to rare epilepsies, which often have many serious and disabling comorbidities, and to patient-centricity and patient-empowerment becoming more mainstream, the epilepsy specialists start talking about these epilepsies in a much boarder way which captures much better the patient and caregiver experience. While years ago most presentations would focus on seizures, at the ECE 2018 we also talked about how to explain families a new diagnosis, the impact on caregivers of having a child with a rare epilepsy, the impact on siblings as well, and many aspects related to intellectual disability or psychiatric comorbidities. This large conversation shift was very refreshing for those coming from the patient side, and is likely to lead to much better patient outcomes.
3. A lot of exciting new therapies are coming, in particular for genetic forms of epilepsy.
Many presentations on the future of epilepsy treatments highlighted the new trials that are coming, most focused on orphan forms of epilepsy. In addition to Epidiolex and fenfluramine (for Dravet and Lennox-Gastaut syndromes) several presentations highlighted ganaxolone from Marinus for treating CDKL5 deficiency disorder, XEN1101 from Xenon for KCNQ2 encephalopathy, and OV935/TAK935 from Ovid Therapeutics and Takeda for multiple rare epilepsies as well.
There are also hopes about future genetic therapies able to increase expression from the heathy gene copy, in all of those cases where the disease is caused by a de novomutation in one of the gene copies. In fact, I had the pleasure to meet with one executive of Stoke Therapeutics attending the conference and hear about their program to use an antisense treatment in Dravet syndrome that will increase the missing protein expression. They are still at a preclinical stage, but from my conversation I came out of the meeting convinced that they are doing an excellent work and that they are well-prepared to take into clinical trials, in some years, what could be the first disease-modifying treatment for Dravet syndrome.
I missed hearing a bit more about gene therapy approaches, which we know are also in development for several of these rare epilepsies where the genetic problem is due to a loss-of-function or a loss-of-expression of a gene. I´m sure it won't be long before these therapies are discussed at epilepsy meetings alongside more classical pharmacological approaches.
4. It is never too late to improve the life of a patient living with a severe epilepsy.
As we talk about those potentially disease-modifying treatments, and at the very least disease-targeting treatments, one hope and one fear emerge. The hope is that they will help us treat not only the epilepsy but also the cognitive, psychiatric, motor and other problems that the patient experiences, since they will target the root of the disease. The fear is that beyond certain age, they might do little for the patient.

I have to thank Prof Sanjay Sisodiya from UCL who works with adults with difficult-to-treat epilepsies for his beautiful and inspiring presentation on diagnosing genetic epilepsies in adult patients. He showed us the image of the medical records of a patient with drug-refractory epilepsy and intellectual disability in his late 50s that was the size of an encyclopaedia collection. Going through that much medical history to try to diagnose the patient would represent an enormous challenge to any physician. The patient’s disease was so severe that he had even stopped talking, so one might think that knowing the specific cause wouldn’t make much of a difference to this patient condition. It turns out the patient had Dravet syndrome, which could be identified in a genetic test, and this finding enabled his doctors to remove the contraindicated medication that he was taking and prescribe instead the standard of care for this particular syndrome. The patient condition improved to the point that Prof Sisodiya showed us a video of the patient, now 66, having a conversation with him.
“It is never too late” was the message to the audience.
It is never too late to diagnose a condition, and it is never too late to offer the patient the best medication that is currently available for their disease. This will be particularly important once we have actual disease-targeting therapies in the market, and only then will we be able to know how much improvement is achieved at each age.
SUMMARY
Overall, an evolution of epilepsy therapy discovery from treating symptoms to treating causes is clear. Much of this is driven by improved understanding of the genetic causes of many types of epilepsy, which points to potential new targets for therapies, and much is driven by the progresses in technologies such as the development of more specific ion channel modulators or antisense strategies that now enable us to tackle those targets.
The second major evolution, linked to the previous one, is a greater focus on rare (orphan) epilepsies over the large symptomatic market. This evolution, as is other disease fields, follows a combination of business advantages and better therapeutic fit, given that many of the rare epilepsies have a genetic cause. For example, as one of the most common genetic forms of epilepsy, Dravet syndrome is likely to be the first --or one of the first-- syndromes to get a disease-modifying treatment using these new approaches.
And last, I have seen another evolution in the way that epileptologists talk about the epilepsies at their major conferences, which has become more holistic and patient-centric. When I started working in the epilepsy therapeutics field 9 years ago, the view of epilepsy by the epilepsy specialists and the pharmaceutical companies was much more focused on the seizures and less on the overall patient experience. Now, as a result of much patient advocacy, physicians look at the disease the same way the patient and their families do. Companies and regulators have also changed the way they used to operate, now including much more directly the patient perspective as part of their work, which has expanded the way they see the disease and think about treatments. And in a medical conference like the ECE 2018, this change was very visible.
I personally missed hearing more about CDKL5 deficiency disorder and other “less popular” rare epilepsies, and about some of the non-pharmacological therapeutics in development, such as antisense therapies and gene therapy approaches. Next stop for the epilepsy community is the American Epilepsy Society meeting starting November 30thin New Orleans where I hope to hear more about those topics, and report back.
Ana Mingorance, PhD
DRAVET SYNDROME DRUG DEVELOPMENT PIPELINE REVIEW 2018
The 2018 Dravet Syndrome Pipeline and Opportunities Review provides a review and analysis of 14 drug candidates in development for the treatment of Dravet syndrome, including 9 products that have received orphan drug designations. It also includes an analysis of the competitive landscape and evaluates current and future opportunities of the Dravet syndrome market.
It has been a year since we released the 2017 Dravet Syndrome Pipeline and Opportunities Review, a market research publication that provides an overview of the global therapeutic landscape of Dravet syndrome.
There have been many developments in the last year, including the approval by the FDA of Epidiolex (cannabidiol) by GW Pharmaceuticals for the treatment of Dravet syndrome, the announcement of positive data from two Phase III clinical trials with ZX008 from Zogenix, and the arrival of a new company pursuing a disease-modifying antisense-based approach (Stoke Therapeutics).
The 2018 Dravet Syndrome Pipeline and Opportunities Review provides a review and analysis of 14 drug candidates in development for the treatment of Dravet syndrome, including 9 products that have received orphan drug designations.
The report includes the most recent updates on Epidiolex (cannabidiol) from GW Pharmaceuticals; ZX008 (fenfluramine) from Zogenix; Translarna (ataluren) from PTC Therapeutics; OV935 (TAK-935) from Ovid Therapeutics and Takeda; EPX-100, EPX-200 and EPX-300 from Epygenix Therapeutics; ZYN002 (transdermal cannabidiol) from Zynerba Pharmaceuticals, BIS-001 (huperzine) from Biscayne Neurotherapeutics; PRAX-330 from Praxis Precision Medicines; SAGE-324 from Sage Therapeutics; OPK88001 from OPKO Health; XEN901 from Xenon Pharmaceuticals; and an ASO program from Stoke Therapeutics.
The 2018 Dravet Syndrome Pipeline and Opportunities Review also includes an analysis of the competitive landscape and evaluates current and future opportunities of the Dravet syndrome market.
The report is now available in this site.
Ana Mingorance PhD

Genes vs syndromes at the International Epilepsy Congress in Barcelona
Should we talk about syndromes based on the gene that causes them or should we talk about them (and treat them) based on the clinical characteristics that they display? Earlier this month, the epilepsy community gathered in Barcelona for the 32nd International Epilepsy Congress and there was a debate between genetic and symptom-base syndrome classification. This debate goes beyond semantics, and has important regulatory and access implications.
THE DEBATE
Earlier this month, the epilepsy community gathered in Barcelona for the 32nd International Epilepsy Congress. There were three interconnected topics that dominated much of the program: genetics of epilepsy, rare epilepsy syndromes and personalized medicine. The other large topic that dominated the agenda was the new epilepsy and seizure classification by the ILAE, and it felt like two separate conferences.
While most scientists are looking inside the cell, looking for genetic changes, many practicing physicians are looking at what they can see, and creating new nomenclatures to define seizure types. And from the patient side this makes a large difference, because the same child with an SCN2A mutation might be diagnosed by a specialist as having SCN2A encephalopathy, and as having Dravet syndrome by another physician based on clinical symptoms.
There has been a trend in the recent years towards creating new syndromes that group patients by the gene that they have affected when their epilepsy is found to have a genetic cause. Some examples of this are CDKL5 deficiency disorder or STXBP1 encephalopathy, which until recently would have be classified as atypical Rett syndrome and Lennox-Gastaut or Ohtahara syndrome respectively, at least in most cases. And that is the catch, that the same gene mutation can lead to different phenotypes in different patients and actually manifest itself as a complete different syndrome.
We see this mismatch between gene and syndrome every day. For example, the nigh before I few to the congress a mother had contacted asking how it was possible that her daughter with Dravet syndrome had exactly the same gene mutation that another patient diagnosed with Ohtahara syndrome had. Then at the congress, Professor Jackie French from NYU presented the case of a family with an inherited mutation in SCN1A that included individuals with mild forms of epilepsies, for example just febrile seizures, but also one child with the much more severe Dravet syndrome. As a physician, she would adjust their medication to their seizure type and treat more aggressively the individual with Dravet syndrome, because their diseases are indeed different ones.
So what is the best classification approach? Should we talk about syndromes based on the gene that causes them or should we talk about them (and treat them) based on the clinical characteristics that they display?
This debate is not just about semantics, it affects a lot how well we develop new medicines and how many patients will benefit from them. So it is important that we get it right.
What follows is my personal view of which syndrome classification is best, when to use it, and what regulatory changes are needed in order to get more medicines to all patients with rare genetic epilepsy syndromes.
OVERLAP BETWEEN SYNDROMES AND GENES
The way I see the debate between genetic and symptom-base syndrome classification is that they are not exclusive; in fact, they form a matrix where many of the epilepsy genes can produce phenotypes that fit into many of the classical (phenotypic) syndrome boxes.
For illustration purposes, I’ve selected a handful of genes and syndromes and created the matrix below. While not all of these genes are found in all of these syndromes, what we have to remember is that many of the epilepsy genes are present across a large number of epilepsy syndromes.

With this in mind, what would be the correct diagnosis for the child of the previous story that had a diagnosis of Ohtahara syndrome before performing genetic testing but was later found to have an SCN1A mutation (typical of Dravet syndrome)? Should the genetic finding change the diagnosis to Dravet syndrome?
I would argue that both a genetic and a clinical diagnosis are needed, so the best diagnosis for this child, if his clinical symptoms are indeed a better match for Ohtahara than for Dravet syndrome, would be “Ohtahara syndrome caused by SCN1A deficiency”. And the other child in the same story would have “Dravet syndrome caused by SCN1A deficiency”. Except for a few truly monogenic syndromes, most epilepsy syndromes have many possible genetic (and sometimes non-genetic) etiologies, and most epilepsy genes produce multiple phenotypes, so I don’t need we need to prioritize the symptomatic classification over the genetic one or vice versa; both are useful and both are necessary. Instead, we should point to the right box in the matrix when diagnosing a patient, which provides both sets of genetic and clinical data.
My purpose when creating this visual matrix is not to break down already rare syndromes into even smaller diseases. On the contrary, the purpose of this exercise is to make the syndrome classification more flexible, so that we can cluster syndromes, and create more flexible indications for developing new drugs and for treating more patients.
REGULATORY IMPLICATIONS
For as long as we didn’t have a good understanding of the genetics behind epilepsy, we have been perfectly OK with symptomatic epilepsy classifications. But the progresses in genetics in the recent decade, and the appearance of many new “genetic epilepsy syndromes”, has opened the door to the development of new drugs that specifically target those genetic defects, creating a new regulatory landscape where our former classification of syndromes based on symptomatology falls short.
The patient groups are already confortable using the genetic syndrome classification, the conference rooms talk more and more about personalized medicine and genetic syndrome classification, and companies pipelines start getting populated with programs that specifically target those abnormal genes and proteins that cause these epilepsies. The next step is to start using the genetic syndrome classification as a suitable drug indication in the drug development process as well.
Let’s go back again to the genes versus (phenotypic) syndromes matrix and see the different therapeutic implications of using one classification versus another.

The first example (scenario A) corresponds to the current preferred way of selecting drug indications. There are 8 different products that have received the Orphan Drug Designation to treat Dravet syndrome. This makes sense because Dravet syndrome has been recognized as a separate clinical syndrome since 1978, way before the genetic mutations that give rise to this phenotype were uncovered. It also makes sense because all of the drugs approved or in clinical trial for Dravet syndrome are symptomatic, meaning that they are not treating any genetic problem.
In short: if the drug treats a particular cluster of symptoms that corresponds to a syndrome, then the best indication for that drug is indeed the classical (phenotypic) syndrome.
But things change when a drug in development specifically targets a disease gene, and this is what is happening right now with a program by OPKO Health designed to increase SCN1A gene expression levels. This product has received the Orphan Drug Designation by the FDA to treat “Dravet syndrome”, because about 80% of patients with Dravet syndrome have mutations or deletions in SCN1A, but would have been more appropriate to match the gene-targeting therapeutic with a genetic patient classification, and target “SCN1A deficiency” as the product indication, like in the scenario B.

If we stick to the classical (phenotypic) syndrome classification for the programs in development that target the gene or protein that are defective in patients, we will be unnecessarily limiting the number of patients that will benefit from that drug. It is clear that in the case of the OPKO program, the indication cannot be simply “Dravet syndrome”, but a subset of patients with Dravet syndrome caused by SCN1A deficiency. But how about the people that having the same SCN1A deficiency have received a different clinical diagnosis based on their symptoms? With the current indication, those people will not have access to such treatment because it will be off-label.
The specific program that OPKO is developing is oligonucleotide-based and therefore very invasive, requiring intrathecal administration, so it is not likely to be used in people with SCN1A mutations and milder phenotypes. But there are small molecule approaches also in development for SCN1A deficiency, and these should not have an indication restricted to a subset of patients with Dravet syndrome. The same will happen with drugs that modulate SCN2A, a related channel that produces multiple phenotypes when mutated and leads to multiple diagnosis. Asking the drug developer to seek approval for only a subset of patients in a rare disease would artificially restrict the number of patients that would benefit from that drug.
This is why we need to get confortable with drugs being developed for a genetic indication that overlaps but doesn’t match the classical (phenotypic) syndrome classification. At the other side of the approval line, physicians must also get confortable with patients having both a genetic and a symptomatic diagnosis so that they can receive both types of medications.
I do believe that indications based on a gene defect (gain-of-function or loss-of-function), when the drug treats the gene or protein that is altered in those patients, will be acceptable and soon become common. What I am more worried about is the need of a scenario C (see next) and the mismatch between the clinical practice and the regulatory pathway.
ADDING COMPLEXITY – SCENARIO C AND MULTI-SYNDROME INDICATIONS
There are drugs that don’t treat the disease (gene or protein) but the symptoms, in the case of epilepsy they treat the seizures, yet they get approved for treating a particular syndrome only. Staying on the example of Dravet syndrome, this would be the case of stiripentol (Diacomit), which is only approved for treating Dravet syndrome but it is simply a GABA-ergic modulator that has no biological reason to work only – or even preferentially – in this syndrome. It is basically a strategic corporate decision to focus on a given orphan indication. Another example is ganaxolone, also a GABA-ergic modulator, that has received Orphan Drug Designations for PCDH19 epilepsy and CDKL5 disorder and is in development for these indications, while there is no obvious biological reason to target those syndromes and no others.
I understand the appeal of orphan indications with limited competition, and how the incentives that the orphan designation brings make these drugs possible so that patients ultimately benefit from them. It is possible that without these incentives these drug developers would have never had the resources to bring forward their drugs to a broader (non-orphan) market. But things start changing when the same developer goes after many orphan epilepsies with a broad symptomatic drug, and we are seeing this right now with Epidiolex (cannabidiol, by GW Pharmaceuticals).
Let me first set one thing straight, I don’t think that GW Pharma is abusing the orphan regulatory route with Epidiolex, they simply have no choice. If they want to develop their product for a large number of patients with orphan refractory epilepsy syndromes, which happen to be separate indications under the traditional classification, they must develop a separate clinical development program for each syndrome. And because they have a broad-spectrum drug with the potential to treat many types of refractory epilepsy syndromes they are running clinical trials for Lennox-Gastaut syndrome, Dravet syndrome, Tuberous Sclerosis Complex and West syndrome. That’s four separate indications.
I wonder how many syndromes should a drug be approved for before it becomes clear that it should have a broader indication and label. In this case, the most appropriate indication would have been “epileptic encephalopathies” or “developmental encephalopathies” or similar, given that the drug appears to behave similarly in all of them. This is what the scenario C is about, drugs that treat a general symptom and should therefore have an indication that is broader than a single individual syndrome (or four).

Recently Takeda started a Phase 2 trial in collaboration with Ovid Therapeutics that is recruiting patients diagnosed with any type of epileptic encephalopathy for treatment with their drug TAK-935. This is what is known as a “basket trial”, where multiple indications are combined. Basket trials started in oncology but this is the first time that we see it run with a mix of epilepsy syndromes. The Takeda trial is not a pivotal trial so it won’t lead to an approval, but it gives me hope that we will one day see this mix of refractory epilepsy syndromes considered as a single indications for drugs that, like the Takeda drug, have mechanisms of action that don’t make them specific for one particular syndrome. As I see it, the basket trial approach should be the future trial design for such drugs, and the broad-label indication should also be the preferred indication unless the product has reasons to only work in a particular syndrome or seizure type.
An interesting consequence of broadening the indication towards this “umbrella” indication might be that it is not longer considered orphan, and therefore the product is no longer eligible for the orphan development pathway and incentives. The answer to this question has important regulatory and commercial implications.
Last, one of the most important aspects of this broad-label scenario C is not the benefit it might bring to drug developers, but the impact it will have towards making sure that all patients with rare epilepsies get access to new medications. Some of the syndromes have patients that can be counted in the low hundreds or even less. The best way to facilitate the development of new medicines for these ultra-rare syndromes is making it possible for them to go together with the other syndromes that share common phenotypes when it comes to evaluating and approving symptomatic drugs. This approach makes medical sense because it is also the same way that physicians work with these syndromes in their regular practice, and it will enable them to stop relying on off-label use of medications.
THE FUTURE
In conclusion, we have a few things to work on when it comes to the debate between genetic and symptom-base syndrome classification:
– First, we need to understand that both classifications are needed because they address different aspects of the disease: the cause and the clinical manifestation.
– Second, as drugs that treat the genes/proteins and not just the symptoms get into clinical trials, we need to be willing to switch from indications that follow the classical (phenotypic) syndrome classification to indications that reflect the drug mechanism so that it can be used across multiple classical syndromes.
– And last, we need to identify better regulatory pathways to make sure that the ultra-rare syndromes can get drugs approved for them as well. In the case of epilepsy this means considering the umbrella indication of epileptic encephalopathies or similar as opposed to artificially slicing indications unless the drug has a syndrome or seizure type-specific mechanism of action.
I hope all of these points will also create a new debate and have an important position in the agenda of the next the International Epilepsy Congress. Until then, I would appreciate your comments and thoughts.
Ana Mingorance PhD
Dravet Syndrome Drug Development Pipeline Review 2017
June 23 is a special day for families of people with Dravet syndrome. It is the International Dravet Syndrome Awareness Day, that in 2017 celebrates its 4th edition. That's why today we announce the publication of the 2017 Dravet Syndrome Pipeline and Opportunities Review, a market research publication that provides an overview of the global therapeutic landscape of Dravet syndrome.
June 23 is a special day for families of people with Dravet syndrome. It is the International Dravet Syndrome Awareness Day, that in 2017 celebrates its 4th edition.
That's why today we announce the publication of the 2017 Dravet Syndrome Pipeline and Opportunities Review, a market research publication that provides an overview of the global therapeutic landscape of Dravet syndrome.
Dravet syndrome is an orphan epilepsy disorder with multiple non-seizure comorbidities and high unmet medical need. The disease has recently gained significant attention as an orphan indication within epilepsy, and as of June 2017, the drug drug development pipeline for Dravet syndrome comprises at least 13 drug candidates, of which 3 are in late-stage, placebo-controlled Phase II or III studies. Two of the products in development are potentially disease-modifying treatments, and 8 different products have received orphan drug designations.
The report includes the most recent updates on Epidiolex (cannabidiol) from GW Pharmaceuticals; ZX008 (fenfluramine) from Zogenix; Translarna (ataluren) from PTC Therapeutics; BIS-001 (huperzine) from Biscayne Neurotherapeutics; OPK88001 (CUR-1916) from OPKO Health; OV935 (TAK-935) from Ovid Therapeutics and Takeda; EPX-100, EPX-200 and EPX-300 from Epygenix Therapeutics; cannabidiol from INSYS Therapeutics; a Nav1.6 inhibitor program from Xenon Pharmaceuticals; and SAGE-324 from Sage Therapeutics.
The 2017 Dravet Syndrome Pipeline and Opportunities Review also includes an analysis of the competitive landscape and evaluates current and future opportunities of the Dravet syndrome market.
The report is now available in this site.
Ana Mingorance PhD

Impatient patients
One day I sent an e-mail that changed my life. During my training as a scientist, I had learned how to study what goes wrong with the brain and to research how we could fix it. Then one day I sent an e-mail to a patient.

One day I sent an e-mail that changed my life. During my training as a scientist, I had learned how to study what goes wrong with the brain and to research how we could fix it. Then one day I sent an e-mail to a patient.
There are about 7,000 diseases that are considered rare because less than 1 in 2,000 people have it. It is not like diabetes, cancer, or Alzheimer’s disease, that we have all heard about. Most people have never heard about these rare diseases, and often not even physicians have heard about them.
Because they are not that common, it often takes many years to find the right diagnosis for these patients, with many left undiagnosed for the rest of their lives. And for the lucky ones that get the right diagnosis, the likelihood of having an approved medication for their disease is 1 in 20. Imagine a 1 in 20 chance of getting a medication for your cancer or your diabetes. A horrible thought, isn’t it? That is the scary world where 350 million people with rare diseases live every day.
But a revolution is changing the way we learn about and treat rare diseases, and it is driven not by progresses in medicine but in technology. Technology, powered by next generation sequencing and bioinformatics, is making the diagnosis of rare diseases much easier and faster. And technology, through the explosion of social media, is helping people with rare diseases and their families connect with other families. And that’s when magic happens.
Five years ago I read an article about a small group of parents that had created a patient organization to find a cure for their children, all diagnosed with a rare neurological condition. They didn’t know how, but they certainly knew what. Andthat is some times all you need to start.
Before finishing the article I sent them an e-mail. I knew about the brain, I knew about drug development, I knew languages and people, and I knew I wanted to help them. That e-mail changed my life. Working with patients changed my life, and not just in the way I now approach drug development and my career. It also changed the way I understand life. Because the only thing harder that being confronted with the reality that there is just a 1 in 20 chance of getting a medication for your disease is when the one with the disease is your child. And many have chosen to get together and do something about it.
That’s why I like to call them impatient patients.
The impatient revolution is already changing the way we do medicine. Patient organizations are becoming central members of the research community and key partners in the development of new treatments. The momentum the impatient patient movement has gained in some fields like rare diseases is unstoppable. And they are not doing it alone. Just like the social media that helped them get started, it is people connecting to people that fuels this movement. Impatient people willing to reach out to patients and their advocates and help create the connections and bridges that they need to succeed. Just like a LinkedIn network, every time we connect with a patient organization we expand their reach, and what starts as a rare disease of few individuals soon becomes a large network of 3rd degree connections that spans across industries and society.
And that’s when magic happens, and why I like to call them impatient patients.
Ana Mingorance PhD
Originally published in LinedIn, October 28 2016