

In February of 2019 I reviewed the state of development of gene therapy approaches for Dravet syndrome at that time. A lot has changed in a year a half, for good. So I felt this update was long due.
Here is a review of the gene therapies in development for treating Dravet syndrome, how each of them works, and the timings that we anticipate for clinical trials.
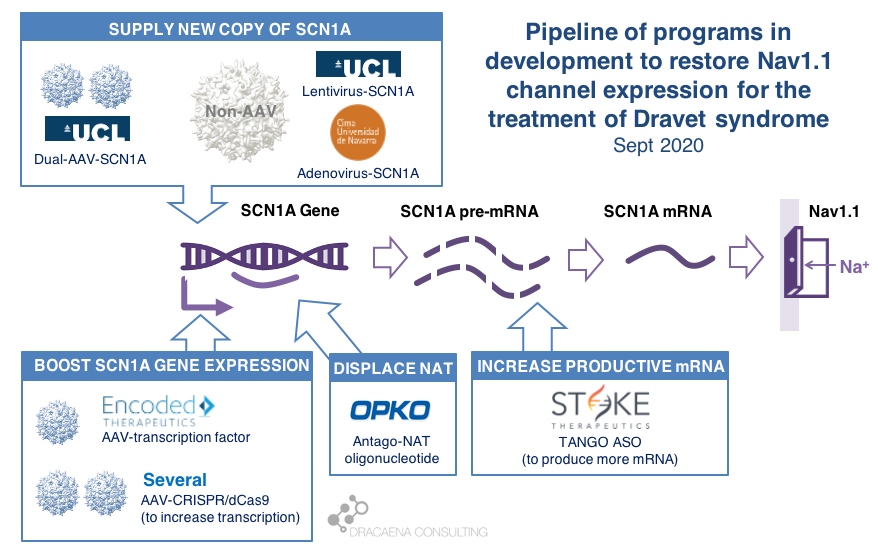
CURRENT GENE THERAPIES IN DEVELOPMENT FOR DRAVET SYNDROME
In diseases like Dravet syndrome where the problem is that a copy of the gene is missing or not functional due to mutations, the desired therapy is one that can restore normal gene expression and therefore normal protein production. In other words, we need more protein.
In the case of Dravet syndrome, the gene is SCN1A, and the protein that is needed is the neuronal sodium channel Nav1.1. As a result of mutations in the gene, the number of Nav1.1 channels at the neuronal surface is not sufficient, there is less sodium crossing the membrane, and the neuron cannot fire properly. The result is Dravet syndrome.
One particularity of Dravet syndrome is that only one of the two copies of the SCN1A gene is affected, the second one is perfectly fine, so that second copy can serve as the supply for extra protein production. As you will see, the most advanced programs are exploiting this possibility.
Broadly speaking, there are two potential approaches to restore protein expression in Dravet syndrome: you either supply the cell with an extra healthy copy the gene, which will lead to more protein being produced, or you try to boost the expression from the healthy gene.
(1) Supply a new copy of SCN1A
When people think about “gene therapy”, the type of therapy they are thinking about is the one where the DNA of a virus gets replaced by the gene that the person needs, and that modified virus is used as a Trojan horse to infect cells and deliver them the therapeutic gene.
The most commonly used virus for gene therapy is the Adeno-Associated Virus (AAV), and because we have so much experience with gene therapies being developed and approved using AAV, this virus is the first choice for most in the gene therapy space. But AAVs are small virus, and have a limited capacity for the size of genes that they can carry, and SCN1A happens to be way too large for using these virus to carry the gene. In January of 2018, before we knew about the new gene therapies currently in development for Dravet syndrome, I reviewed this problem in the article “big gene, small virus”.
One way around this is to use other viruses that are larger and can therefore carry larger genes inside. One of these is the Adenovirus, which we all hear much about recently because it is also the virus of choice for the AztraZeneca vaccine in development for COVID-19.
|| The Spain-France-Israel consortium CureDravet is developing a gene therapy for Dravet syndrome using Adenovirus, a type of high-capacity virus that is large enough to contain the entire SCN1A gene. The leading Spanish group from Rubén Hernández has presented early results at conferences (e.g. European Paediatric Neurology Society (EPNS) 2019) showing rescue of multiple disease phenotypes in transgenic mice by administration of an adenovirus-based SCN1A gene therapy. As of the last public update this is still a preclinical program working towards the development of a clinical candidate.
Another way around the gene-vs-virus size issue is to use not one, but two AAVs, and to make each carry half of the SCN1A gene. This is a strategy that is also being explored for gene editing using CRISPR because the CRISPR approach requires expressing in the tissue proteins whose genes are too large to fit into an AAV. So the strategy of two AAVs is being explored both for rare genetic diseases with large genes such as Dravet syndrome, and for gene editing in general.
|| At UCL, the team of Rajvinder Karda and Simon Waddington is working on two approaches. One is to use another type of large-capacity virus, Lentivirus, to carry the SCN1A gene. The second approach uses two AAV virus, each containing half of the SCN1A gene, which are able to recreate the full channel once they co-infect the same cells. As of the last public updates these are also still preclinical programs working towards the development of clinical candidates.
(2) Boost expression of SCN1A
Another strategy to restore Nav1.1 levels is to target the good SCN1A gene copy or some stem downstream of it to increase protein production, without needing to add an external gene copy with a virus. This one is the strategy most advanced for Dravet syndrome.
There are several ways to do this, and luckily for us many of these approaches are being pursued and some of them are already in clinical development or getting very close.
As a small biology primer: genes (like SCN1A) are large stretches of DNA that contain the information needed to produce proteins (like Nav1.1). These large stretches of DNA are first copied into large stretches of RNA (pre-messenger RNA or pre-mRNA), then some sections that are not meant to be part of the final protein are removed from the RNA to make it shorter (mRNA), and then the mRNA is read to produce the final Nav1.1 protein.
What happens in Dravet syndrome as a result of having a mutation (or deletion) in one copy of the SCN1A is the following:

By now we know that the levels of Nav1.1 protein production are regulated in the cell at different levels along this sequence of steps, offering scientists multiple points where to act when trying to boost production of Nav1.1:
Regulation at the step of gene expression 1: the SCN1A gene has sequences around it that tell the cell how much the gene should be copied and therefore how much RNA should be produced. These are promoter or enhancer or other regulatory regions that could be targeted to boost gene expression and protein production.
Regulation at the step of gene expression 2: cells produce some RNAs that are not meant to produce proteins, but to match the sequence of some genes and prevent them from being read by the cell. SCN1A happens to be the target of one of those Natural Antisense Transcripts, or NAT. This endogenous “repressor” of SCN1A expression could be also targeted to boost gene expression and protein production.
Regulation at the step of RNA processing: neurons produce a large amount of pre-mRNA of SCN1A, with only a fraction of it being processed into mRNA which is the final form that will be used to produce proteins. This creates a “reserve pool” of pre-mRNA that could potentially also be targeted to get more productive mature mRNA and therefore boost protein production.

The first company to try one of these approaches was OPKO Health. OPKO decided to go after the Natural Antisense Transcripts that limits SCN1A expression by developing an oligonucleotide that could displace it, releasing the good SCN1A copy and leading to more mRNA and more Nav1.1 protein levels. Because their oligonucleotide antagonizes the NAT for SCN1A they called it an AntagoNAT (OPK88001, previously CUR-1916). While OPKO indicated plans to initiate clinical trials as early as in 2017, these timelines were moved to 2018 and then 2019 and as of summer 2020 there are no news of whether this program is still active. In the meantime, other corporate programs have appeared and progressed further into the clinic.
As a side note: oligonucleotide therapeutics are NOT gene therapies, if we stick to the actual meaning of gene therapies using genes as therapies. But because oligonucleotide therapeutics target gene expression or mRNA processing leading to more protein production we often bundle them together with the “real gene therapies”. Both are approaches that target the genetic problem in the disease, either correcting it or compensating for it (like when boosting the healthy gene copy expression). Both are expected to result in increased levels of Nav1.1. And both are expected to lead to transformational improvements across different symptom domains in patients. So for all purposes it makes sense to talk about together when reviewing gene therapies.
2018 brought the good news that Stoke Therapeutics was developing an antisense oligonucleotide treatment to boost expression of SCN1A as well. This oligonucleotide binds to the pre-mRNA, facilitating the transformation of some of the pre-mRNA “reserve pool” into mature mRNA and leading to more Nav1.1 protein. Stoke recently published their preclinical proof of concept (July 2020 and August 2020), showing how they were able to produce an antisense oligonucleotide able to trigger this pre-mRNA to mRNA processing specifically for SCN1A and not affecting other sequence-related sodium channel genes. Their therapeutic candidate, called STK-001, increased expression of the SCN1A mRNA and Nav1.1 protein in mice, and had remarkable efficacy in mice with Dravet syndrome caused by SCN1A haploinsufficiency.
|| Since my early 2019 update, Stoke Therapeutics had an IPO, obtained an FDA Orphan Drug Designation for STK-001, and in August of 2020 it initiated a Phase 1/2s study in patients 2 to 18 years with Dravet syndrome. The dosing of the first patient in this study represented a historical moment for the Dravet syndrome field: for the first time, a person born with Dravet syndrome was given a therapy designed to correct their genetic problem (SCN1A haploinsufficiency). That day the field moved from developing and testing symptomatic treatments to developing and testing disease-targeting treatments, which the patient community often prefers to call cures.
The program from Stoke Therapeutics is the most advanced of all gene therapies and gene therapy-like approaches in development for Dravet syndrome. The second program in line is a gene therapy approach by Encoded Therapeutics that was still in stealth mode in 2018.
|| In June 2019, Encoded Therapeutics emerged from stealth mode with a series C of $104M with a lead program for Dravet syndrome. Encoded managed to beat the gene-vs-virus size problem by using AAV, the gold-standard viral vector for gene therapy, to deliver to the brain a synthetic transcription factor for SCN1A. In other words, the SCN1A gene might be too large, but we can still AAV to deliver to the brain the gene of a specific SCN1A booster. In barely one year, Encoded has closed another funding round of $135M, obtained the Orphan Drug Designation and Rare Pediatric Disease Designation by the FDA for their ETX101 gene therapy for Dravet syndrome, and anticipates to start clinical trials in 2021.
Several academic efforts follow Stoke and Encoded in the pursue of boosting expression from the good SCN1A gene copy. All of these projects are in early preclinical stages, and they have not yet published a solid proof of concept in a Dravet syndrome mouse model, which is an initial stage prior to advancing the treatments towards clinical trials. These programs are therefore all years away from clinical trial initiation, with no guarantee of succeeding.
|| An academic group in Italy, with funding from CURE and the Dravet Syndrome European Federation, is researching an oligonucleotide approach to boost production of Nav1.1 protein though a combination of transcriptional and translational activity. There have been no recent public updates on the progress of this project.
|| The Vania Broccoli lab in Italy in collaboration with Gabriele Lignani lab at UCL are using a CRISPR approach to target the promoter of SCN1A and increase its activity, therefore boosting production of Nav1.1 from the good SCN1A gene copy. They use a variant of CRISPR that uses dead Cas9 to find – but not to cut – the desired genomic region and activate it. They have demonstrated the ability of upregulating Nav1.1 using this system using lentivirus in vitro, and published an early proof of principle in mice by using two AAVs to deliver all of the different elements of the CRISPR system into the brain, which would not otherwise fit into one AAV. It is early to know if these dual-AAV approaches will achieve sufficient biodistribution and expression in the brain, or if single-AAV or single-Adenovirus approaches are needed, probably reducing the size of the cargo like Encoded is doing with their transcription factor approach.
|| The Yamakawa lab in Japan has also published a recent study using CRISPR with dead Cas9 to increase SCN1A gene expression and protein production in mice. For multiple reasons, their approach cannot be considered a proof of concept for the future clinical treatment. For example they used a type of AAV (PHP.B), which has fantastic biodistribution in mice but not in primates, and administered it via intravenous administration. Peripheral administration of AAV to target neurological diseases also seems an approach less viable in patients than in mice. In this case part of the CRISPR system was genetically encoded in the mice, so they only needed administration of one AAV. This would also not be possible in patients, who would need the entire treatment to be delivered to them using suitable route of administration and virus type. But what this study provides is a proof of biologic rescue of SCN1A expression in GABAergic neurons starting at 4 weeks of age in mice, and the improvement in seizures, mortality and behavioral phenotypes that result from such rescue.
|| SCN1A encodes for the alpha subunit of Nav1.1, so less alpha subunit means less mature sodium channels, but there is also a beta subunit involved in channel formation which is encoded by SCN1B. The Hampson lab in Toronto recently published a gene therapy approach using AAV to deliver an additional copy of SCN1B, which is a small gene that fits the virus. The reasoning is that upregulating the beta subunit might be able to drag more alpha subunits to the surface and result in more total Nav1.1. In the study, funded by Dravet Canada and the US Dravet Syndrome Foundation, mice heterozygous for SCN1A had improvements in mortality, seizures and behavioral outcomes, although some of these were more pronounced in males or females. None of the SCN1A boosting approaches had reported any sex-bias when it comes to efficacy. The beta subunit produced by SCN1B is auxiliary to multiple channels, not unique to Nav1.1, and the study did not determine which of these channels might be mediating efficacy, so the relevance of this study to the SCN1A-targeted therapeutics is unclear.
FINAL THOUGHTS
There are several differences between oligonucleotide treatments and gene therapies. Some of these are differences that are more important for us scientists. For example to better understand the need to increase SCN1A expression in all cells that naturally express the gene (what oligonucleotides do) versus the convenience of boosting it only in those that are the most affected in the disease (an option available only for gene therapies because they can use cell-specific promoters). Other differences are more important to patient families, such as the need to repeat dosing with oligonucleotide treatments, usually spaced out by several months, versus the once-and-done single-administration approach of the gene therapies. In the end what we all want to see is which approach produces the most clinical improvement with the least side effects and burden of administration, and it is very likely that we will want to have multiple options in the market.
When thinking about these options, the field of SMA comes to mind. Spinraza (by Biogen) is an ASO able to increase the production of SMN, and a total game-changer in a terrible disease that used to kill babies. Then Zolgensma (by Novartis) reached the market, as the first gene therapy using AAV to deliver a copy of the SMN1 gene. With both in the market we have started seeing clinical studies to evaluate the combination of the ASO and the gene therapy in patients with SMA. And last month FDA approved risdiplam (Evrysdi) from Genentech/Roche which is a small molecule that can be taken orally and that increases the production of SMN. In 4 years the field of SMA has gone from facing a death sentence to having three very different modalities all able to rescue the missing protein expression (ensuring access to these treatments is a different topic, but I still want to highlight that there is a lot of work to do there).
So while we keep an eye on Stoke and Encoded and the development of their two very different approaches, we might want to keep another eye following the developing of small molecule activators of the Nav1.1 channel, for which there are several programs in the development.
We also want to make sure that the current oligonucleotide treatments and gene therapies in development are successful. A major challenge they face is that we don’t know how to design a clinical trial with a gene therapy in Dravet syndrome. So far clinical trials measuring seizure frequency have been very successful, but a gene therapy is expected to improve the syndrome beyond just seizure frequency. The field of Dravet syndrome is still immature when it comes to clinical outcome measure development and validation for non-seizure outcomes (for non-scientists in the audience: we don’t know how to quantify improvements of the disease in a clinical trial beyond seizures). Both Stoke and Encoded are running observational studies that will hopefully identify the best outcome measures and endpoints that will be needed for pivotal studies.
And another development that I miss is that despite so many approaches trying to increase the levels of Nav1.1 we don’t have any biomarker that could help us measure the levels of functional or total Nav1.1 in patients. This will make it hard to interpret clinical trials where a dose of the treatment is ineffective based on the selected outcome measures. And having a biomarker for protein levels will provide early data to encourage longer patient monitoring as we wait for some of those outcome measures to show enough of a change. So in parallel to a race for developing new treatments, we are seeing a race to de-risk the field fast enough so that those treatments get their best shot at succeeding in a pivotal trial.
IN SUMMARY
There are multiple gene therapy and oligonucleotide programs in development for Dravet syndrome including those that supply and extra copy of the SCN1A gene and those that boost expression from the healthy SCN1A gene copy.
Clinical trials have already started, with Stoke Therapeutics initiating the first clinical trial with a disease-targeting therapy in Dravet syndrome in summer 2020.
Behind Stoke, gene therapies are approaching the clinic with Encoded Therapeutics having the most advanced clinical candidate and preparing for trials in 2021.
I look forward to seeing precompetitive collaborations around the common challenges of validating clinical outcome measures and biomarkers, which are needed to maximize the success of gene therapies for Dravet syndrome.
Ana Mingorance PhD