

Epilepsy Insights

REPASO DEL FORO CDKL5 2022
La octava edición del Foro CDKL5 tuvo lugar en Boston, los días 7 y 8 de noviembre. El Foro es una reunión anual que organiza la Fundación Loulou y en la que científicos y miembros de la industria farmacéutica se reúnen con representantes de la comunidad de pacientes para repasar los últimos avances en el campo.
Este es un repaso para los grupos de pacientes de las principales novedades del Foro CDKL5 2022.
Hace ya ocho años que la Fundación Loulou organiza una reunión anual, el Foro CDKL5, donde los científicos de academia y de industria trabajando en el síndrome de deficiencia en CDKL5 (CDD), junto con representantes de los grupos de pacientes, se reúnen para compartir las últimas novedades y avanzar hacia tratamientos y una cura.

La edición de 2022 tuvo lugar los días 7 y 8 de noviembre en Boston, volviendo al formato presencial tras dos años en formato virtual debido a la pandemia. Eso ha hecho el Foro de 2022 aún más especial. Para mi este ha sido el mejor Foro desde 2018 que fui al primero. Ha habido un progreso enorme en lo que sabemos de CDKL5 y la velocidad de progreso de las terapias, así que voy a resumir las principales noticias del Foro de 2022 y compararlas con cómo estaban las cosas la última vez que nos reunimos en persona, en 2019 también en Boston.
1. EL AÑO EN QUE NOS UNIMOS AL CLUB DEL 5 PORCIENTO
Seguro que habéis oído eso de “hay más de siete mil enfermedades raras, pero solo el 5% tiene algún tratamiento aprobado”. Es un club especial ese 5%, son las enfermedades raras que han recibido suficiente atención e inversión para que se haya desarrollado y llevado a ensayos clínicos un tratamiento específicamente para ellos.
En 2022, CDD se unió a ese club.
En 2022 ganaxolona se aprobó en Estados Unidos para tratar las crisis epilépticas en CDD, y estamos a esperas de las decisión de la agencia europea que se espera en cuestión de meses. Cuando nos vimos por última vez en Boston en 2019 hablábamos de que Marinus estaba cerca de completar el reclutamiento para su ensayo clínico de fase 3 con ganaxolona en CDD. Desde entonces Marinus completó el reclutamiento en un ensayo por primera vez global en CDD, en medio de una pandemia también global, y ya el fármaco está aprobado y disponible para los pacientes en Estados Unidos y esperamos que pronto en otros países. Hay que destacar la valentía del equipo de Marinus de apostar por una enfermedad que no tenía tratamientos previos, y de luchar a través de una pandemia para sacar a mercado la que sería la primera terapia para CDD. Han allanado el camino que ahora es más fácil para los que vienen detrás.
Y resulta que la aprobación del primer fármaco para CDD solo es una de las dos grandes noticias del año…
2. LA TERAPIA GÉNICA PARA CDD AVANZA A ENSAYOS CLÍNICOS
Una de las mayores esperanzas para las familias CDKL5 es la llegada de las terapias génicas a ensayos clínicos. En CDD, a la mitad de las neuronas (en niñas) o a todas (en niños) les falta CDKL5 por tener un gen mutado. Y eso les impide producir la proteína CDKL5 que es muy importante para el funcionamiento neuronal. Las terapias génicas buscan usar un virus para llevar una nueva copia del gen CDKL5 a las neuronas, y que así puedan volver a producir la proteína [ver el repaso de estrategias de tratamiento en la sección siguiente].
En el Foro 2022, Emil Kakkis, que es el director ejecutivo de Ultragenyx, por fin dijo las palábras por las que llevábamos esperando los últimos años: “la terapia génica para CDD viene a ensayos clínicos, creemos que el año que viene”.

Más tarde vimos también la ponencia de Sharyl Fyffe-Maricich que es quien lidera el proyecto de la terapia génica de CDKL5 en Ultragenyx, y nos explicó todos los pasos que han tomado para asegurarse de que su terapia génica llega a tantas neuronas como sea posible antes de sentarse con la agencia reguladora a hablar de empezar ensayos clínicos. La calidad de la investigación que hace esta gente es impresionante, y Emil nos explicó que el desarrollo de una terapia a través de ensayos clínicos siempre es difícil pero que estamos juntos en todo esto.
La última vez que nos vimos en Boston en 2019 vimos la presentación de datos de terapias génicas en ratones con deficiencia en CDKL5, y en ese momento nos hacíamos aún dos preguntas muy importantes: si llegarían a funcionar suficientemente bien como para avanzar a ensayos clínicos, y cuando sería. Ahora por fin ya tenemos respuestas y fechas orientativas: si todo va bien, los primeros ensayos con la primera terapia génica para deficiencia CDKL5 podrían empezar en 2023.
Esta es la segunda de las grandes noticias del año, y sin duda el notición del Foro.
3. ATACANDO A CDKL5 POR TODOS LOS LADOS
Durante la cena de recepción del Foro, Phil Railly (un descubridor de terapias para enfermedades raras muy importante) nos compartía varias historias de enfermedades que se consideraban intratables hasta que padres de niños afectados tomaron cartas en el asunto y jugaron un papel clave para desarrollar curas. Su mensaje era que “no hay enfermedades intratables”.
Luego al día siguiente Emil Kakkis nos recordaba que “CDD tiene aspectos reversibles, y eso no pasa en muchas otras enfermedades neurológicas”. Hacía referencia a lo que hoy en día sabemos de que en ratones con deficiencia en CDKL5 la enfermedad es reversible, y también sabemos que no es una enfermedad degenerativa, ni siquiera de las típicas de neurodesarrollo donde la falta de la proteína durante etapas claves de desarrollo causa daños irreversibles. Hoy entendemos que la deficiencia en CDKL5 es un desorden de “mantenimiento de función neuronal”, como vimos en el Foro de 2020, con lo que hay mucha esperanza de cara a la posibilidad de devolver la expresión de CDKL5 a los pacientes con CDD.
En el Foro 2022 vimos actualizaciones sobre muchas aproximaciones terapéuticas para atacar a la deficiencia en CDKL5 por todos los lados. Os hago un resumen de lo que vimos:

Tratar los síntomas: el ensayo con fenfluramina está en marcha y es el segundo ensayo de Fase 3 que tenemos en CDD. Fenfluramina ha tenido resultados muy buenos en el síndrome de Dravet, y la empresa que la desarrolla (Zogenix) ha sido comprada por una de las mayores empresas farmacéuticas líder en el campo de epilepsia, UCB Pharma. Los ensayos clínicos están reclutando en Estados Unidos y pendientes de arrancar en Europa y Japón.
Tratar los síntomas: Takeda evaluó la eficacia y tolerabilidad de soticlestato en un estudio pequeño en CDD, y nos trajo una actualización de 8 de esos 12 niños que se quedaron tomando el fármaco tras terminar el ensayo. Todos han experimentado mejoría en el control de crisis y los padres reportan mejoras importantes en otros dominios como atención y comunicación, con lo que esperamos que Takeda considere CDD como otra indicación para soticlestato.
Corregir los problemas en las otras proteínas: si sabemos qué proteína no funciona bien al faltar CDKL5 podríamos ir directamente a arreglarlas y compensar por la falta de CDKL5. En este punto hubo un descubrimiento muy importante este año, al que dedico la sección siguiente entera.
Aportar más proteína: el desarrollo de terapias de reemplazo enzimático para CDD (cuando produces la proteína en el laboratorio para dársela a los pacientes) ha resultado ser tremendamente difícil. Maria Luisa Tutino presentó en el Foro su trabajo para conseguir la producción de proteína CDKL5 funcional en el laboratorio para usarla como terapia, y los desafíos con los que se iba encontrando y que van solucionando uno tras otro. Yo espero que se unan más investigadores y empresas a intentar conseguir una terapia de reemplazo enzimático para CDD porque hace mucha falta y es muy complejo.
Arreglar el ARN mensajero: el gen CDKL5 se copia en ARN mensajero, y este a su vez sirve de instrucción para hacer la proteína CDKL5. Hay varias estrategias para intentar sacar proteína funcional a partir de un ARN mutado, y en el Foro vimos una estrategia en Boston para hacer oligonucleótidos antisentido (ASOs) que es un tipo de terapia disponible en otras enfermedades genéticas. Estos ASOs servirían para ciertas mutaciones en CDKL5, no todas, por ejemplo para intentar leer el ARN cuando hay mutaciones de “splicing” o si hay algún cacho de CDKL5 menos importante, haciendo que la célula se salte la lectura de ese trozo del ARN (salto de exón). Hay ejemplos de estas estrategias en la atrofia muscular espinal y en Duchenne, pero para CDKL5 apenas están empezando.
Reactivar la segunda copia del gen CDKL5: tuvimos dos presentaciones de grupos que buscan usar una especie de CRISPR para llegar a la copia inactiva de CDKL5 (en niñas) y hacer que se exprese. El invesitgador Kyle Fink lleva años liderando uno de esos proyectos y le acaban de conceder cerca de un millón y medio de dólares para avanzar su terapia génica para CDD (que hace que se lea la segunda copia). Esta año recibió el premio al mejor Laboratorio del Año en el Foro CDKL5.
Arreglar el gen mutado: hay varias estrategias en desarrollo para corregir las letras en un gen, o para poner una letra que falta. Majid Jafar, co-fundador de la Fundación Loulou, nos explicaba que a Loulou le falta una letra en el gen CDKL5 y que al saber el diagnóstico su madre se preguntaba “¿y por qué no le ponen la letra que falta y ya está?”. Pues el investigador David Liu está usando también una especie de CRISPR que se llama “prime editing” para hacer justamente eso, poner las letras que faltan. Nos enseñó cómo ya pueden arreglar el gen CDKL5 de niños como Loulou en células en el laboratorio, y actualmente están trabajando en hacer que funcione en ratones. Este tipo de edición genética está empezando ensayos clínicos en otras enfermedades y se descubrió hace apenas 5 años. El objetivo de David no es ya solo poner o quitar una letra, sino como nos dijo “cambiar el gen entero por una copia buena”. Las presentaciones que da este hombre son siempre una ventana hacia el futuro.
Aportar una copia nueva del gen: esta es la terapia génica clásica, en la que los científicos usan un virus (generalmente uno llamado AAV) para llevar el gen que falta a las neuronas. Ese virus no se multiplica, sino que sirve para llevar el gen a las neuronas y que a partir de ese día ya puedan producir la proteína, en este caso la de CDKL5. En el Foro 2022 tuvimos el notición de Ultragenyx de que su terapia ahora avanza hacia la fase de ensayos clínicos.
4. UN DESCUBRIMIENTO MUY IMPORTANTE PARA ENTENDER CDKL5
Vimos muchas presentaciones en el Foro sobre entender mejor cómo funciona la proteína CDKL5 y las consecuencias de su falta para el cerebro. Pero solo voy a hablar de una en este resumen, porque es un descubrimiento fantástico que ayuda a entender lo que hace la proteína y lo que pasa en la enfermedad. También es una puerta abierta a diseñar nuevos tratamientos.
El descubrimiento nos llega del laboratorio en Londres de Sila Ultanir. La proteína CDKL5 es una kinasa, que son proteínas que lo que hacen es modular la función de muchos otros tipos de proteína. Esto multiplica los problemas, porque cuando falta la enzima tienes también todas esas proteínas que no funcionan bien. El grupo de Sila ha descubierto una proteína muy importante de esas que necesitan a CDKL5 para funcionar bien, y resulta ser un canal de calcio (Cav2.3). Cuando falta CDKL5, ese canal de calcio de abre bien pero luego se cierra muy despacio, lo que lleva a demasiado calcio pasando por el canal.
Estos es muy importante porque muchos de los síndromes de neurodesarrollo con epilepsia están causados por problemas con canales iónicos, las llamamos las canalopatías. Por ejemplo el síndrome de Dravet es por falta de canales de sodio, y la epilepsia por KCNQ2 es por mutaciones que dan demasiada actividad a canales de potasio. Curiosamente hasta hay un síndrome ultra-raro causado por tener el canal de calcio Cav2.3 demasiado abierto, y eso es justamente lo que han visto que pasa también cuando falta CDKL5. Esto era inesperado, pero explica muchas cosas.
Y digo que es un descubrimiento muy importante por dos razones:
1) Muchas veces nos preguntamos… si CDD está causado por faltar una kinasa por qué se parece tanto a algunas canalopatías. Ahora sabemos la respuesta: ¡CDD es parcialmente una canalopatía!
2) Este descubrimiento abre las puertas a diseñar nuevos tratamientos dirigidos a corregir las consecuencias de la falta de CDKL5. Ahora sabemos que queremos que alguna empresa desarrolle un fármaco bloqueante de Cav2.3.
5. MUCHO PROGRESO AMPLIANDO LA CAJA DE HERRAMIENTAS DE INVESTIGACIÓN
Para poder entender qué hace CDKL5 y probar terapias antes de que lleguen a ensayos, los científicos necesitan modelos de la enfermedad en animales o en células en cultivo. Y ha habido un esfuerzo de titanes por parte de investigadores en muchos países para encontrar animales o tipos de cultivo celular al que la ausencia de CDKL5 le provoque síntomas claros (lo que llamamos fenotipos).
Y en investigación casi siempre se usan ratones y ratas modificados genéticamente para tener el mismo problema que la enfermedad en humanos. Pero este año vimos presentaciones de equipos que están ampliando la caja de herramientas de investigación de solo ratones y ratas a incluir muchos otros, como peces, moscas, renacuajos y hasta cerdos con deficiencia en CDKL5.

Vimos ponencias de dos equipos usando peces cebra como modelo de deficiencia en CDKL5, uno de Portugal y uno de Boston. Los peces tienen algunos de los problemas que vemos en personas afectadas, por ejemplo problemas óseos, y están buscando hacer peces a los que les falte CDKL5 solo en la mitad de las neuronas que es lo que pasa en niñas y por tanto es la mayoría de los casos.
Mis presentaciones favoritas sobre modelos de deficiencia en CDKL5 fueron dos:
El laboratorio de Ibo Galindo, en Valencia, ha hecho moscas de la fruta (o mosca del vinagre) con deficiencia en CDKL5 y visto que desarrollan epilepsia y su enfermedad es tan severa que hasta viven menos que las moscas sin modificar. Lo curioso es que las moscas solo tienen un gen CDKL, en vez de cinco como las personas, con lo que a estas moscas ¡les falta el equivalente de CDKL1/2/3/4 y 5! Y les produce los mismos síntomas que tienen moscas de otras epilepsias genéticas, las de CDKL5 incluso tienen cambios en su comportamiento. Las moscas de la fruta son de mucha utilidad como herramienta de investigación genética, porque se le pueden quitar y poner genes y ver su función, o en este caso darles los genes humanos de los cinco CDKL y ver cuales les corrigen la enfermedad.
La empresa Vyant está usando células madre de niños con CDKL5 para producir organoides. Los organoides son como una pelota de neuronas y otras células cerebrales todas producidas a partir de las células madre que se sacan de la piel o muestra de sangre con lo que llevan el ADN de los niños, y que forman en cultivo como un mini-cerebro (en la foto son la bola que está a la derecha del todo). Pues resulta que los organoides de niños con CDKL5 tienen la versión in vitro de epilepsia, que son descargas eléctricas hipersincronizadas. Y lo están usando como plataforma de cribado de fármacos y ya han encontrado algunos compuestos.
Lo que más me impresiona es la tenacidad de tantos científicos haciendo un esfuerzo de gigantes para no dejar piedra sin remover, mirando en cada animal y cada tipo de cultivo celular y buscando la forma de encontrarle la señal de la falta de CDKL5. Gracias a eso hoy tenemos un abanico de modelos animales y celulares que permiten hacer muchas preguntas científicas, tanto de biología como de tratamientos, y este tipo de trabajo requiere un gran esfuerzo de grupo, no lo puede hacer un laboratorio solo. Y de eso trata la sección siguiente de este resumen.
6. “ESTAMOS EN ESTO JUNTOS”
Hay un mensaje que se convirtió en el lema del Foro 2022, y es “estamos en esto todos juntos”. Lo oímos en palabras pero también lo vimos en acciones. Estos son solo algunos de los ejemplos que vimos durante el Foro:

Que no se quede nadie atrás. Hay personas con CDD en todos los países, y eso implica también que son personas con todos los idiomas. Por desgracia la barrera lingüística es una de las principales barreras a la hora de llegar a la comunidad internacional de pacientes. Para poder reunir datos de tantos afectados por CDD como sea posible, y poder mantenerles informados de noticias como ensayos clínicos, el registro CDKL5 Registry está disponible desde hace poco en la mayoría de los grandes idiomas: Inglés, Español, Francés, Alemán, Italiano, Árabe, dos versiones de Chino, Japonés y Ruso. Ha sido un trabajazo pero estamos en esto todos juntos.
Empresas colaborando en estudios clínicos. Cuando nos vimos la última vez en Boston en 2019 lanzamos la propuesta de que hacía falta hacer un estudio clínico en CDD para validar escalas clínicas (para verificar que sabemos medir los síntomas de CDD de la forma que hace falta para ensayos clínicos). Y lo que pedíamos no era simple, porque nuestra propuesta es que fuera solo un estudio en el que colaboraran todas las empresas, co-diseñándolo y ejecutándolo juntas, para así no duplicar (o quintuplicar) esfuerzos y sobrecargar a las familias. Este año Xavier Liogier, de la Fundación Loulou, nos hablaba desde ese mismo escenario pero esta vez para anunciar que ya ha entrado el primer paciente a ese estudio clínico, llamado CANDID, que incluye muchos países incluida España. Entre esas dos ponencias ha habido muchísimo trabajo, incluso llevamos el estudio a examen por la agencia americana del medicamento, y también ha supuesto un importante paso hacia la colaboración por parte de las 7 empresas que se juntaron para hacer el estudio posible. Estamos en esto todos juntos.
Colaboraciones preclínicas. La comunidad de investigadores en CDKL5 siempre ha sido excepcionalmente colaborativa, y el Foro tiene una serie de sesiones de grupo en la que nos coordinamos. Este año salió la propuesta de que las empresas que están buscando terapias para CDKL5 se unan a esas colaboraciones preclínicas (de investigación en animales) porque suelen estar presentes en la conversación pero lo que no hacen es abrir sus libros de laboratorio y enseñarnos los datos. Lo que se busca es que los investigadores trabajando con ratones con CDD en universidades y en empresas se puedan sentar juntos y comparar resultados y poder así validar los mejores síntomas en ratones que podemos usar para investigar (los síntomas más robustos y mas reproducibles entre laboratorios). Los grupos académicos ya lo hacen y el objetivo es que se unan los de las empresas. Espero que el año que viene en el Foro 2023 podamos confirmar que hemos abierto la colaboración y que se han unido a un esfuerzo tan necesario, porque estamos en esto todos juntos.
La Alianza internacional ayuda a acelerar estudios clínicos. Una dificultad habitual de los ensayos clínicos en enfermedades raras es que les cuesta mucho a las empresas identificar los hospitales que hacen falta. La Alianza Internacional de CDKL5 ha compilado una lista de los hospitales de cada país de la alianza en los que los miembros tienen médicos de confianza con experiencia en CDD. En el Foro aprendimos que esta lista ha facilitado y acelerado la identificación de hospitales para el estudio CANDID, y espero que también acelere los futuros ensayos clínicos. Estamos en esto todos juntos.
El liderazgo de las familias CDKL5. El Foro CDKL5 es una conferencia médica y científica, donde nos reunimos para repasar los últimos avances y para organizarnos de cara a seguir avanzando. Pero también se trata de un congreso liderado por los padres de una niña con CDD, donde las familias tienen un papel prominente tanto en la agenda como en las conversaciones. Abrimos el Foro con las palabras de Natalie Ladly, de CDKL5 Canada, que compartió con todos nosotros lo que supone para su familia vivir con CDD (#brynnstrong!). Luego durante la cena de gala Simon y Fiona Walsh de CDKL5 Irlanda también nos abrieron las puertas de su casa y su familia, y para cerrar el congreso Rick Upp, de la IFCR, nos hizo un resumen de lo que ha pasado este último año en la comunidad de pacientes. El premio al “Paladín del Progreso” que da el Foro cada año a las personas que más impacto han tenido en CDKL5, fue este año a Antonino Caridi, de CDKL5 Italia, uno de los líderes mas querido y respetado de la comunidad de pacientes en su lucha por una cura para CDD. Y tuvimos representantes de pacientes de 14 países (¡ya hay más de 30 en la Alianza!) que participaron en el Foro, mezclándose con investigadores y participando de manera activa en las conversaciones, sobretodo durante las sesiones de debate. Es así que a través de sus palabras y sus acciones las familias CDKL5 nos enseñaron durante el foro que estamos en esto todos juntos.

RESUMEN
Si se puede resumir el Foro de 2022 en unas pocas frases, sería:
No hay enfermedades intratables.
CDD es ahora parte del 5% de enfermedades raras con un tratamiento aprobado.
La terapia génica viene a ensayos clínicos.
Y desarrollo de una terapia a través de ensayos clínicos siempre es difícil pero estamos en esto todos juntos.
La Fundación Loulou fue fundada en 2015 con la misión de tener “tratamientos para CDD en ensayos en 2020, y curas en ensayos en 2025”. En aquellas fechas no había ni un solo fármaco en desarrollo para CDD, pero si que había fe en la ciencia y en el poder de crear una comunidad en torno a una enfermedad rara. Tras las noticias del Foro 2022 ahora ya sabemos que es realista pensar que incluso antes de 2025 habrá personas que hoy viven con CDD que habrán recibido una terapia génica como parte de esos ensayos clínicos. Este ha sido sin duda muy buen año para la investigación en CDKL5.
Espero que os haya gustado el resumen. Ya me diréis lo que os parece en los comentarios.
Ana Mingorance, PhD
Nota: este texto captura mis impresiones de las presentaciones del Foro que más me interesaron como científico y como defensora de los pacientes, no es un texto oficial del congreso emitido por la Fundación Loulou. Escribo estos resúmenes para los padres de personas con CDD, así que a veces me tomo ciertas licencias a la hora de explicar las partes mas técnicas.

Top 5 insights from the American Epilepsy Society meeting (2021)
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. These are my top 5 insights from the American Epilepsy Society 2021 meeting.
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. I look forward every year to the first week of December for this reason.
I look for therapies for rare genetic epilepsies, and in the recent years this area has exploded to take over much of the AES meeting. Because of that, I will focus this update on the rare genetic epilepsies, or Developmental and Epileptic Encephalopathies (DEEs). There is a lot more that is presented at the AES meeting and that I will not cover. These are my top 5 insights from the American Epilepsy Society 2021 meeting.
1. Natural History Studies are tremendously important
When companies develop treatments for seizures as a symptom, it is straightforward to run trials: just count seizures. But when companies start developing treatments that treat the cause of that epilepsy, and in particular when we are talking about complex neurodevelopmental syndromes with epilepsy, then clinical trials are more complex than just counting seizures, and need more solid data on the usual symptoms and severity that those patients have so that we can know if there is efficacy. This is where Natural History Studies (a type of observational study) specifically designed to inform on clinical trial design come into play. And we are seeing a big number of these.
Several of these ongoing studies were presented at the AES meeting. The BUTTERFLY study by Stoke Therapeutics is an observational study to characterize clinical scales to measure the non-seizure aspects of Dravet syndrome, and has already shown promise with several scales that could be used in interventional clinical trials. The ENVISION study by Encoded Therapeutics, has also helped validate scales and determine the baseline characteristics on non-seizure outcomes in young children with Dravet syndrome. Both companies are developing treatments to rescue SCN1A gene expression in Dravet syndrome, so these observational studies are designed to make those treatment trials possible, and both observational studies are already producing very useful data.
We also saw example of more proactive observational studies that are already ongoing even before companies are ready for trials, like an SCN8A Natural History Study resulting from a partnership between hospitals and patient groups, and a consortium working on STXBP1 Natural History. While these studies are simpler than the company-run Dravet syndrome studies, and don’t validate scales, they help us understand the collection of symptoms and their severity in these syndromes, and guide the design of the clinical-trial enabling studies. It is also remarkable that both studies include data from more than 500 patients from each syndrome!
If you are running or supporting a rare epilepsy patient group, I recommend you also check these two additional resources to understand why these studies are so important and consider how it could work for your disorder:
1) The pre-competitive industry collaboration involving seven biopharmaceutical companies and the Loulou Foundation to run an observational study specifically designed to evaluate the feasibility and suitability of a collection of clinical outcome measures in CDKL5 deficiency: here and here.
2) The amazing Mike Granglia from SYNGAP Research Fund explains in a recent video why Natural History Studies specifically designed to inform on clinical trial design are so so important to not delay the initiation of clinical trials for these syndromes. Please watch it, the Natural History Studies part starts around minute 5:45 but the entire videos are fantastic.
2 – Gene therapies are coming for many epilepsies
There are many reasons to believe that many of the genetic epilepsies will benefit much from restoring gene expression in patients, even in older individuals. And we saw several studies in mice presented at AES where scientists returned gene expression to mice with genetic epilepsies showing very good efficacy. See for example this study in CDKL5 Deficiency Disorder, this one in SCN1A Dravet syndrome, and this one in STXBP1-related DEE.
To be able to do that in patients, scientists need to develop gene therapy approaches using antisense oligonucleotides (ASOs) or virus that can bring to the brain the missing gene, or virus that can bring to the gene CRISPR approaches or transcription factors (see review of options for Dravet syndrome here). We saw several of this presented at AES this year.
The most advanced program is the one from Stoke Therapeutics. In addition to presenting an update about their observational study, Stoke presented new information about how their ASO to increase SCN1A distributes within the brain in non-human primates, and the likely doses that are needed to produce enough sodium channel in the brain in trials. From their data, it seems that repeated administration of 30mg of STK-001 (their ASO to increase SCN1A levels) will be a good target to to achieve active brain levels in patients. They are not yet administering those levels to patients because the interventional trial is still at early stages, and it is a Phase 1/2 trial without any prior trial in healthy volunteers, therefore starting with only one administration of STK-001 to Dravet patients at very low dose and moving from there to higher doses, still with only one administration to monitor safety, and then starting with repeated dosing also from lower concentrations to higher. From this early data, Stoke reports good tolerability, and a trend towards having less seizures but all participants had received either only one administration of STK-001 or multiple administrations of 20mg, so we really have to wait for the trial to reach higher dose with repeated administration in order to see the therapeutic potential of this treatment. We might potentially see this first from their UK study, called ADMIRAL, where Stoke will evaluate multiple doses of up to 70mg [see Note for parents at the end of this text].
Behind STK-001, there are several therapies in development, all designed to increase SCN1A for Dravet syndrome, that have not yet started clinical trials. You can see a summary of all those approaches here, where I explain the biology of SCN1A deficiency and the options to increase it.
Encoded Therapeutics is developing ETX101, a gene therapy that uses an AAV virus to upregulate SCN1A in the brain. They are preparing to start clinical trials, and this year presented data on their observational study as well as on understanding caregiver perspective to better design their upcoming clinical trial. A company called CAMP4 has acquired the rights to the ASO program from OPKO to upregulate SCN1A, and talked about the preclinical data that is available for this program at AES. And there are at least three academic groups developing viral-based gene therapies for Dravet syndrome, all focused on increasing the SCN1A gene: the adenovirus program that we saw last year at AES, a new one this year, and a CRISPR-ON approach being developed in Italy.
We also saw preclinical data for a gene therapy program for SLC13a5 Deficiency, showing efficacy in young mice and also in young adults, which is very promising towards future studies. And scientists at UCL in the UK are working on bringing gene therapy to non-genetic epilepsy! Their gene therapy is designed to sense if neurons are hyperactive, turn on and produce potassium channels (which makes neurons less excitable), and as soon as neurons are not hyperactive anymore, turn off. This means that neurons would only make use of that gene therapy when they truly need it. They are still trying it in mice, so it is at early stages, but I found this research one of the most groundbreaking presented AES.
3 – Not all is gene therapy: Many exciting progresses with small molecule drugs
We had a very good year of progresses in small molecule drugs (“normal medicines”) for epilepsy, including for rare and non-rare epilepsies.
Probably the most impressive is the Phase 2 data of XEN1101 in adult patients with focal onset seizures, which is the most common seizure type. There are more than 20 drugs approved for treating focal onset seizures, and trials are done with the new drug added on top of whatever the patients are already taking, so it is very difficult to see impressive efficacy in this oversaturated space. Yet XEN1101 achieved a reduction in seizures of up to 52% at the highest dose. This is rare and exceptionally good.
There were also several updates on late-stage programs presented. We saw post-Phase 3 trial data for fenfluramine in Dravet syndrome, and for ganaxolone in CDKL5 Deficiency Disorder, both looking really good. Ganaxolone has also shown efficacy in PCDH19 epilepsy in a Phase 2 study, with 61,5% of seizure reduction. There is an ongoing trial in young children with diazepam nasal spray as a rescue medication, and Takeda presented how they plan to count seizures in Lennox-Gastaut syndrome in their Phase 3 trial with soticlestat which has a novel glutamatergic mechanism of action.
And I liked to see several innovative compounds coming up in the earlier pipeline. Eisai has a GAT1 inhibitor called E2730 as a potential alternative to tiagabine. A KCC2 activator might have potential for treating seizures in Rett syndrome. Praxis has a sodium channel blocker called PRAX-562 with preclinical efficacy for SCN8A and SCN2A Gain-of-Function epilepsies, and Xenon has sodium channel potentiators with preclinical efficacy for Dravet syndrome (SCN1A).
I might be missing some compounds, but it is clear that 2021 was a very good year for the developments of medications for epilepsy, with some really strong data in clinical trials and in the “real world”, and with new drugs that have novel mechanisms of action at early discovery stages.
4 – Understanding genetics in epilepsy
It is likely that most epilepsies have some genetic contribution. As we learn more about it, and genetic testing becomes more common, we will find many more individuals with rare genetic syndromes and start finding sub-groups of patients with “common” epilepsy that might respond better to certain treatments.
Xenon Pharma has a drug in Phase 3 for KCNQ2-related epilepsy, and they are one of the sponsors of the Invitae genetic testing program for epilepsy. So they interrogated the data from Invitae to see how many of the cases are due to KCNQ2 mutations. I knew this disease was one of the most common genetic epilepsies, but I did not expect the numbers that they got: KCNQ2 is by far the leading cause of neonatal epilepsy, and still the most common in children under 6 months of age. We urgently need early genetic testing for epilepsy to not miss these cases!
There are still new genetic causes of childhood epilepsy being found, like SLC7A3 and SYNJ1 mutations, and scientists are able to create mouse models of genetic epilepsies that require mutations in more than one gene. And a large collaboration of scientists has put together a portal called NDD-CNV to collect information on copy number variations in neurodevelopmental disorders (many with epilepsy) to assist with variant interpretation, which is very important for syndromes like Dup15q and 8p disorders.
And we saw several genotype/phenotype studies where scientists try to see if different types of mutations produce different prognosis which is a common question from parents after genetic diagnosis (see for example SCN1A and STXBP1). There is a little bit of correlation when you compare mutation type in hundreds of patients, but it doesn’t seem that we will be able to predict the development of a child just knowing the specific mutation that they have in a given gene. There was also a massive study in 11,500 patients with epilepsy that relied on phenotype keywords to see if they could be matched to genes, and it identified certain keywords that are preferentially associated with some syndromes like “hemiclonic” and Dravet syndrome, and that suggests that we could potentially use good phenotypic descriptions to prioritize genes to test in these patients. I still believe the best option is to just sequence all people with epilepsy for all epilepsy genes or the entire exome/genome, with particular urgency for young children.
5. Patient groups are building research-enabling platforms
This year we saw some very important research efforts led by patient groups, with the purpose of making it very easy for scientists and companies to work in their disorders:
The NDD-CNV portal is hosted by the Broad Institute and co-developed with the Dup15q Alliance, Project 8p, and Ring 14 US and Ring14 International. It is a natural synergy among structural variant disorders (as opposed to point mutations) and I encourage you to check out the link.
A similar effort, the GRIN Portal, is also hosted by the Broad Institute and developed in partnership with several national and international GRIN patient organizations, and provides information about the genes, the disorders that they cause when mutated and the consequences of specific variants to facilitate interpretation.
Yet another portal hosted by the Broad Institute is the SLC6A1 Portal, with analogous information about SLC6A1 and also with collaboration from patient organizations.
Shout out to Dr Dennis Lal from the Cleveland Clinic and the Broad Institute for making those three portals positive (and more, like the sodium channel portal), and being such an amazing advocate and promotor for patient-led research in genetic epilepsy.
The SYNGAP Research Fund and Ciitizen have also brought together the largest cohort of SYNGAP1-related disorder patients in a platform to collect real world evidence, which will help design clinical trials for this syndrome. And I already mentioned the SCN8A Natural History Study and STXBP1 Natural History Study which are also happening thanks to the effort and leadership of patient organizations.
I believe this type of effort is the best investment that patient groups can do, more than investing on any single research program. It requires that the patient group takes a leadership position in the field and builds the blocks that will facilitate and de-risk individual programs. And it has required a cultural change where clinicians and academic scientists are now very open to partnering with patient organizations and co-leading these efforts. 2021 was a great example of this.
LOOKING INTO 2022
As we look into the new year there are some planned milestones that I look forward to, and some things that I hope will also come true:
I look forward to efficacy data from STK-001 in Dravet syndrome, and the initiation of clinical trials with ETX101.
I look forward to ganaxolone getting approved for CDD, and fenfluramine showing positive results in the ongoing Phase 3 (if interim data is already available by then)
I look forward for more genetic therapies moving into trials. There are several preclinical programs with ASOs and viral-based gene therapies, and in the coming years we should see several of these moving into trials.
And I look forward to seeing progress with several small molecules that target disease-causing proteins, as in KCNQ2, SCN2A, SCN9A and SCN1A Dravet syndrome. Short of gene therapies these might be the best treatments for these disorders.
Ana Mingorance, PhD
Note for parents of someone with Dravet syndrome: when fenfluramine and CBD started trials in Dravet they already had data from adults with other diseases, same for soticlestat and others, so by the time they started trials in Dravet they could immediately administer the drug at the therapeutic dose daily for three months to see efficacy. That is different with STK-001 because it is a disease-specific treatment targeted to people with deficiency in SCN1A so they had to start trials directly in patients and figure out the tolerable and effective dose also in patients, going little by little and starting with low doses. The way I see it, it wouldn’t be correct to compare the current Phase 1/2 Stoke trial with a Phase 3 with other drugs. They are still building up to getting to the best doses, and so far so good.
Disclaimer: These are my own impressions from the presentations and topics that I was most interested in. I write these texts with the parents of individuals with rare epilepsies in mind, so excuse also my lack of technical accuracy in parts.

MAIN LESSONS FROM THE 2021 CDKL5 FORUM
For the past seven years the Loulou Foundation hosts an annual meeting where scientists and drug developers working on CDKL5 deficiency, together with representatives from patient organizations, meet to discuss the latest advances.
Here are the main news and take-home messages from the 2021 CDKL5 Forum that took place in November 1-2 2021.
For the past seven years, the Loulou Foundation has hosted an annual meeting, the CDKL5 Forum, where scientists and drug developers working on CDKL5 Deficiency Disorder (CDD), together with representatives from patient organizations, meet to discuss the latest advances in the field. You can find written summaries from the past three meetings here: 2018, 2019 and 2020.
The 2021 CDKL5 Forum edition took place November 1-2, and for the second year, due to the ongoing pandemic, it was held fully on-line. The Loulou Foundation turned this into an opportunity to also host a series of workshops prior to the 2-day conference, turning the Forum into a giant Think Tank. This made this year’s Forum quite unique, and I hope the pre-conference workshops are here to stay.
I know that many in the CDD patient community are waiting for this written summary, so I will try to extract the main conclusions from the Forum with this audience in mind. But I want to start with a warning: if you are reading this summary mainly looking for an answer to “when are the gene therapy trials for CDD starting?”, the best answer that we have today is “we don’t know yet”. I understand this might feel disappointing to some, as if we are not progressing fast enough, and potentially make you feel a bit hopeless. That’s only human, so I want to acknowledge that upfront.
In the next paragraphs I will walk you through the magnitude of the effort and the tangible progress that has taken place around research and treatments for CDD, and that was presented at the Forum. I invite you to read it in the spirit of celebrating each step and each progress and small victory along the way, because we have come a very long way, and this year the progress in biology understanding and therapy development has been particularly significant.
It is remarkable how far we’ve come in the last few years, and the size and commitment of the community of scientists and companies that has grown around CDD.
Because of that, I have structured the summary of this year’s Forum around the following themes: hope, tangible progresses, strategy and partnerships.
1. HOPE
The 2021 Forum started with the voice of the patient. This year it was IFCR’s President Karen Utley, who shared with the audience the life of her daughter Samantha, and the impact that CDD has on Samantha and her entire family. Samantha sees 14 different specialists a year and takes 15 different medications, which gives us some quantitative data for the complexity of this disorder. From the many great messages from Karen, I keep two: (i) small improvements can be life changing for patients and families, so please don’t focus only on cures and keep working to make more smiles and happy days, and to (ii) please keep hope alive for those who are feeling too overwhelmed to stay hopeful.
Her plead to keep hope alive was adopted as a motto by many of the conference speakers in the two days that followed.
There were several moments during the conference where science showed us there are many reasons for hope. These are some of those reasons:
Hope because mice show us that CDD is a good candidate for genetic rescue

Just two weeks before the Forum we celebrated the publication of a landmark study showing that CDD is reversible in mice (article link). Zaolan (Joe) Zhou, from the University of Pennsylvania, also presented the results of his study at the Forum, which show that CDKL5 is needed throughout life to keep proper neuronal functioning and that, in mice, turning “on” the CDKL5 gene in near-adult mice that grew up with CDKL5 Deficiency leads to large reversal of most symptoms. This is very valuable because it helps us learn what is biologically possible, and that CDD is not just a disease of neurodevelopment but also of neuronal maintenance.
In layman terms, it appears that in CDKL5 Deficiency, the brain developed fine and the problem is that neurons are not talking to each other correctly. So once we put the protein (or the gene) back they can start talking to each other correctly, because nothing was really lost: there was no neuronal death or failure to form those neurons, and they are not in the wrong location, which are all things that happen in other neurodevelopmental diseases. This study is a big reason for hope, and Joe received the CDKL5 Forum Award for Lab of the Year for this contribution to the field.
Hope because other diseases are already seeing great results
As part of the gene therapy session, Krystof Bankiewicz, from Ohio State University, showed us videos of children with AADC Deficiency who participated in a clinical trial for a gene therapy to deliver to their brains the AADC enzyme that they are missing. If you saw the videos you probably had chills. Patients with AADC Deficiency have very strong hypotonia, intellectual disability, dysautonomia and other complications due to missing an important enzyme necessary for producing dopamine and serotonin. Prior to treatment, the mobility of these patients is so impaired that they reminded me to several of the boys with CDD, unable to even hold their heads up. In the videos, we saw children go from that extreme hypotonia state to seeing changes 1 month after gene therapy, sitting after 6 months, walking with assistance of DAFOs after a year, and totally unrecognizable walking by themselves after 18 months. Krystof explained that many of the patients also became verbal, which was highly unexpected. While we don’t know yet how fast and how much people with CDD will improve once we have a gene therapy for them, seeing the results in this other enzyme deficiency help us learn what is biologically possible, and it is a big reason for hope.
Hope because science (and scientists!) keep coming up with great genetic tricks very quickly
While we are today very focused on gene therapies to bring an extra copy of CDKL5 to each neuron in the brain, we got to see at the Forum some of the science that is coming afterwards, and that will allow us to fix the mutated gene in patients with genetic diseases. David Liu, from the Broad Institute, showed us how he is using an advanced version of the CRISPR system to correct mutations where one letter is missing, and showed us how they can do it already in cultured cells from a girl with CDD (this is still all very early and being optimized). And Feng Zhang, also from the Broad Institute, showed us another advanced version of the CRISPR system that instead of correcting one letter in the DNA can swap large sections of a gene or an entire gene. So we can imagine one day getting a single gene therapy that gets to the brain and swaps the entire CDKL5 gene for a good copy, and that could potentially be used for kids with all types of mutations.
But before we get to see these gene editing approaches turned into actual treatments, we have many other therapies coming our away. I review those in our next section: tangible progress.
2. TANGIBLE PROGRESS
Progress in three gene therapies
Last year at the 2020 Forum we talked about the two gene therapies for CDD that had shown to have efficacy in mice deficient in CDKL5, or as we call it in science had “preclinical Proof-of-Concept”. We also talked about how gene therapies need to go through three stages: first prove that you can treat a mouse, then prove that you can use it in larger brains (non-human primates) since it is much more difficult than in small mouse brains, and then finally start the first trials in a very small group of patients to determine safety before exposing more patients.
This year we saw not two, but three gene therapies, and how the most advanced ones are now working on that second stage that goes between showing that you can treat mice and being able to start clinical trials.
The gene therapy that we saw presented for the first time is the program between the University of Pennsylvania (Penn) and the company Amicus, and it is a very interesting hybrid between gene therapy and protein (enzyme replacement) therapy. They call the approach cross-correction, because the therapy uses a virus to bring to neurons a copy of the CDKL5 gene (this is a gene therapy), with a special version of CDKL5 that will produce a secreted protein. The idea is that the neurons that are successfully corrected will act as local sources of enzyme to supply to neighboring cells. This was the first time that we saw mouse data from this project at the Forum, and the researchers are trying to see how much more efficacy they can get by using this special version of CDKL5 as opposed to plain CDKL5.
For the other two gene therapies, from Penn (in collaboration with Elaaj Bio) and Ultragenyx, we got to see some of the work that goes into getting the therapies ready for patients now that we know both of them work in mice.
The gene therapy program from Penn/Elaaj came with new EEG data, showing that it is not only the symptoms but also the EEG abnormalities in mice with CDKL5 deficiency that get corrected with the gene therapy. And more importantly, they showed two newer versions of the gene therapy, more advanced than the one we saw last year, and how with each iteration they are getting better and better distribution in the non-human primate brains. They are making these upgrades trying to get as much expression of CDKL5 and as broad as possible, and as the presenter said, “we are getting closer to the target of robust expression in the brain” that they want to see before moving into clinical trials.
The team from Ultragenyx also dedicated most of their presentation to talk about those next steps after seeing efficacy in mice with CDD (which we saw last year). They told us about the need to fully evaluate safety and toxicology in animals before entering the clinic, and also about all of the additional work that they are doing to prepare for clinical trials involving natural history and biomarker data. This was very impressive, and showed how Ultragenyx has many people in the clinical and regulatory teams and many other teams in addition to their preclinical researchers all working on the CDKL5 program.
So we don’t know yet when trials might start, but we know that these three programs keep getting closer and closer to the clinic, and that we have some of the best people in medicine working on solving all of the difficult barriers that need to be overcome to take these treatments into clinical trials (like better brain distribution, and development of biomarkers, and clarity on clinical scales to use). You will see more of this in the last section about strategy and partnerships.
Progress with ganaxolone
If everything goes well, ganaxolone will become the first drug to be approved for the treatment of CDD. The FDA plans to make a decision in March 2022, and the EMA some months later. In the meantime, Marinus has an Expanded Access Program ongoing which is now also open to Europe.
What I found really exciting from the Marinus presentation was data from their open-label studies, where they showed how patients from the Phase 3 trials are doing as they rolled over into the open-label extension. Since it is also openly available in their website and not confidential, I am going to copy their slide here because the data looks really good and it is better seen in the graph:

In the solid green bars, you see the patients that received ganaxolone during the Phase 3 study, and then continued taking it in the open-label extension. Remember that the study is blind, so no one knows if they are taking placebo or ganaxolone during the 17-week study. These patients had a seizure reduction of 30% at the end of the trial, and for the following 12 months their seizure control got better, staying more around 40-45% seizure reduction. This is not trivial, because it is common for patients with CDD to respond to a new medication for 3 or 4 months and then lose efficacy, while ganaxolone doesn’t show signs of losing any efficacy after a year, all the contrary.
And in the striped green bars you see the patients that received placebo during the Phase 3 study, but then started taking it in the open-label extension. Because the study is blind no one knew during the 17-week study if they were getting the drug and not responding to it or if they were in placebo. On average this group had seizure reduction of only 6,9% during the trial (since they were not on the drug), and as they rolled over into the open-label extension their seizure control got better and better until they caught up with the ones that we already taking ganaxolone in the trial.
This is a beautiful graph, that shows that ganaxolone has better efficacy in CDD than what is seen only during the double-blind phase, and that you have to give it enough time to show its full potential. It is also quite encouraging to see how stable the efficacy of ganaxolone remains over time.
Progress with fenfluramine
Fenfluramine is an anti-seizure drug that is already approved for Dravet syndrome where it has exceptionally high efficacy in reducing seizures. After a successful small pilot study in CDD run at NYU, the company Zogenix has been working on preparing for a Phase 3 trial of fenfluramine in CDD.

And the Zogenix team gave us the great news at the Forum that the Phase 3 trial with fenfluramine in CDD has already started and is already enrolling patients in the US clinical sites! They are enrolling children and adults with CDD as young as 2 years old, and once they have enough safety data they might open it to 1-year-olds and older, and patients need to have active epilepsy since that is what the study is measuring. For the trial, Zogenix is looking for 100 participants.
This is really good news because it means that at least 100 patients with CDD will get to try fenfluramine, and if it has in them even a fraction of the efficacy that we have seen in Dravet syndrome and in the small study in CDD, it will be very meaningful for those patients and their families.
3. STRATEGY AND PARTNERSHIPS
The Forum becomes a Think Tank
During the week prior to the conference, the Loulou Foundation hosted focused workshops that covered four areas of priority for the development of therapies for CDD:
Spontaneous and induced seizure activity in CDKL5 knockout animal models
New technologies for improved therapeutic cargo delivery in the central nervous system
Biobanking of patient samples and fluid biomarker discovery/validation to support CDD therapeutic development
Functional biomarker development/validation and inclusion in clinical development plans
For each workshop, the Foundation invited a group of academic, industry and clinical researchers, and for some also patient representatives, to try to map out (i) where we are, (ii) where we need to be, (iii) what are the technologies or fields that we should be paying attention to, and (iv) how we can get organized to address those challenges including specific proposals for collaborations. This is what turned the 2021 Forum edition into a true Think Tank, and the outcomes from each of the workshops were presented during the main conference to share with the entire community.
To give you an example for how these workshops worked, when talking about new technologies for delivering genes to the brain, we covered all approaches from engineered viruses to synthetic biology, nanoparticles and the possibility to attach CDKL5 to proteins that cross the blood-brain-barrier to use them as shuttles. And we had scientists from companies that are developing all of these approaches sit on the same workshop and share with everyone else their thoughts and proposals. This level of collaboration among traditional competitors is highly unusual, and it is one of the themes that we kept seeing during the conference: the building of a scientific community around CDD that includes many companies joining forces to help us get to better treatments. You will hear more of this later, when I talk about the CANDID study.
The long road to solving the translational puzzle
Translational medicine refers to the bridge between treating mice and treating people. It seeks to answer questions such as: “how will we know that we have increased CDKL5 protein in the brain?”, “how will we know if we are improving synaptic function?” and “how are we going to measure improvements in patients in the trial?”.
To answer these questions, we need to develop ways to see things, which can be biomarkers (like things you can see in a blood test, or brain scan) or clinical scales. Because CDD is a relatively newly-described disease, we still don’t have these biomarkers and scales.
The good news is that there are many collaborative efforts ongoing to address all these questions and develop all these biomarkers and outcome measures. This was a central theme of this year’s Forum and the subject of two of the pre-meeting workshops, with important presentations on these topics:
Need and plans for a biobank of patient samples, with standardization of analytical techniques and data sharing including universities and companies
EEG-based biomarkers, presented by Eric Marsh from CHOP, as a way to measure dysfunction of brain activity in mice and patients with CDD, to detect potential restoration with treatments
Blood-based biomarkers, presented by Max Bianchi, from the Trinity College Dublin, as another way to measure dysfunction of brain activity in mice and patients with CDD, to detect potential restoration with treatments
A US and Australia NIH-funded study coordinated by Tim Benke from the University of Colorado to also test and validate biomarkers as well as new clinical scales for CDD in an observational study
An international observational study coordinated by the Loulou Foundation in partnership with 7 pharma companies to test and validate clinical scales for CDD, called the CANDID study (see also next section)
In addition to helping us solve the translational puzzle, what all of these efforts and studies have in common is that they all require the active participation of the patient community. This could look like organizing the collection of some blood samples at your next national families meeting to then send them to Dublin, or participating in an EEG study in Philadelphia or Boston, or volunteering to participate in the observational studies that will take place in clinics from around the world. You will likely hear of some study near you!
So if you have a family member with CDD, I encourage you to consider participating in some of the studies as well as enrolling into the CDKL5 Registry (this one only needs data, no samples or visits to a clinic needed).
We cannot answer all of these key translational questions without you.
A first-of-its-kind partnership
Just a week before the Forum, the Loulou Foundation announced the launch of a pre-competitive industry collaboration involving seven biopharmaceutical companies together with the Loulou Foundation. The consortium will run an observational study specifically designed to evaluate the feasibility and suitability of a collection of clinical outcome measures in people with CDD from all ages.
Xavier Liogier, from the Loulou Foundation, explained how this study fits into a larger roadmap for preparing the CDD field for the arrival of better therapies and cures, and that already started with the PFDD meeting in 2019 and getting an ICD-10 code for CDD.
Observational studies are like a clinical trial but instead of testing a drug you are testing the clinical scales to check that they are good for that disease. These studies are very important to be ready for clinical trials that look at several symptoms or disease domains. Usually, when companies run observational studies of this kind, they do it alone, so in diseases where there are many companies developing therapies we might end up seeing several observational studies, each one run by a different company with a slightly different design.
We wanted to avoid this for CDD.
The idea for the CANDID study was born two years ago as a big hairy audacious goal: what if we could get all of the companies working on CDD come together, and co-design a single observational study that would meet everyone’s needs, get also the FDA to review it while we are at it, and then run that single study with all of the companies working together to minimize the number of observational studies that we need in CDD.
It sounded good. And all of the clinical teams at the different companies that we talked to also told us that it would be something they would support. That’s what gave us the conviction that we needed to try to make it happen. The problem was that we had no reference for other collaborations of this type. There was no book that we could read to know how to do this. This was going to be a first-of-its-kind partnership.
So for the past two years we had to figure out a way to co-design a study among so many parties without the study dying of decision paralysis, and we had to figure out the best way to build a consortium to govern the study where all companies would be equal partners. We also had to grow as a Foundation to be able to lead and coordinate these efforts, which brought us new team members like Xavier Liogier, who has the ideal human and professional mix to lead this study, and new strategic partners in clinical operations to help run this observational study with the very same standards as a pharma-run Phase 3 trial.

This is why I am so proud to be part of this story, as part of the Loulou Foundation team that made the CANDID study possible. We knew that all the companies working on CDD would like to see a consortium-run study, and we all understood that we needed to try to avoid having multiple corporate observational studies in CDD. So we figured out how to make this first-of-its-kind partnership happen, and in the process, wrote the first chapter of that book.
The CANDID study is about to start and will look for 100 participants of all ages (with active epilepsy or not), hoping to include study sites in the US, Canada, UK, France, Spain, Italy, Germany and Russia. This will help us make the study accessible to the large majority of patients diagnosed with CDD.
Strategy and partnership also on the advocacy side
Natalie Ladly, secretary for the International CDKL5 Alliance, helped close the conference with a presentation about the Alliance and a call for action.
There are 17 patient organizations under the Alliance (check them out at https://cdkl5alliance.org/) and one of the top priorities is to help the community be trial-ready. Natalie explained that they have 47 clinical trial sites around the world, at least one on each of the countries under the Alliance, and made a call to the industry and the medical community to reach out to the national patient groups and the Alliance and tell them what would help them (industry and clinicians) the most. Essentially, “help us help you”, and “by the way we have solved your problem for finding trial sites”.
I believe every year I write a longer summary. There is just so much going on, and so many news and progresses in many areas, that it is difficult to know what to cut from the summary. My main impressions from the 2021 CDKL5 Forum are that we are seeing solid tangible progresses in many areas, and that we seem to be turning a corner, where the ratio of individual labs working on CDD versus partnerships is changing and now a majority is working on the same agenda to solve the same problems together. This partnership model was started by the academic and clinical centers, and it has now also reached biotech and pharma companies.
This is why I want to commend in particular the professionals from so many biotech and pharma companies (we had 24 companies at the Forum!) that came to the Forum and sat down with their traditional competitors to share their best ideas for CDD and agree on joint efforts to make these happen. And I believe that the industry consortium behind the CANDID study is a testimony to that spirit of breaking previous barriers to industry collaboration, not just by saying nice words, but by working together to make progress actually happen.
And that is why hope, tangible progresses, strategy, and partnerships, where the themes of the 2021 CDKL5 Forum.
I hope you enjoyed this summary. Please let me know your thoughts in the comments.
Ana Mingorance, PhD
Disclaimer: These are my own impressions from the presentations and topics that I was most interested in as a scientist and patient advocate, and not an official text about the Forum by the Loulou Foundation. I write these texts with the parents of individuals with CDD in mind, so excuse also my lack of technical accuracy in parts.

CDKL5 DEFICIENCY IS REVERSIBLE
CDKL5 deficiency is biologically reversible, bringing much hope and creating a sense of urgency for the development of restorative treatments for CDKL5 deficiency disorder (CDD). These are the conclusions of a publication from Zhaoland (Joe) Zhou’s lab from the University of Pennsylvania. The study was just published in the Journal of Clinical Investigation, and it is poised to become a landmark study in the field.
CDKL5 deficiency is biologically reversible, bringing much hope and creating a sense of urgency for the development of restorative treatments for CDKL5 deficiency disorder (CDD).

These are the conclusions of a publication from Zhaolan (Joe) Zhou’s lab from the University of Pennsylvania. The study was just published in the Journal of Clinical Investigation, and it is poised to become a landmark study in the field.
It has been almost 15 years since a similar publication from Adrian Bird in mice with Rett syndrome due to mutations in MECP2. Bird’s lab showed that Rett syndrome was reversible if the gene was returned to adult mice who had developed without MECP2. The study told us that it is likely never too late to rescue MECP2 deficiency, and it encouraged research into the genetic rescue of Rett syndrome in patients. The experiment (and the investigator) became so famous, that gene therapy researchers from universities and industry have been asking for the last 15 years “has anyone already done the Adrian Bird experiment for this gene?” for any genetic syndrome that they consider working on.
Joe Zhou and his colleagues just did that for CDKL5. And the response is affirmative: CDKL5 deficiency is biologically reversible.
Here is a summary of these findings, and what they mean in the context of developing curative treatments for CDD. There is also a press release from Penn here.
WHAT THE PUBLICATION SHOWS, IN LAYMAN’S TERMS
I love the elegance of this study, and I am impressed by the amount of work and exquisite diligence that went into creating this publication. This is why I feel so strongly about the importance of these results.
Terzic et al (2021) creates two types of genetically modified mice. One type is mice that are born with normal CDKL5 expression, but that can stop expressing CDKL5 when the investigator decides. The other type are born with CDKL5 deficiency, but can produce CDKL5 when the investigator decides. Essentially there is one mouse strain where scientists can decide when the disease will start, and one where scientists can decide when it will stop. And they use these mice to answer key questions about the biology behind CDKL5 deficiency.
Because the protein CDKL5 is expressed in the brain throughout life, the scientists hypothesized that perhaps it is important also throughout life, not just in early development. This is not trivial. People born with a bad copy of CDKL5 develop a disease called CDKL5 deficiency disorder (CDD) that starts very very early in life, leading to global developmental delay in addition to seizures and other complications. Because it starts so early, it looks like having enough CDKL5 is important for very early development, and maybe if you make it past all those critical early stages of post-natal development with normal expression of CDKL5 then you will always be ok even if you stop producing enough CDKL5 later in life. Short answer: you are not.
It is never too late to develop CDD. That is why the protein CDKL5 is expressed in the brain throughout life, because it is always needed.

This is what the publication shows, by letting mice develop normally and then turning off their CDKL5 gene when they are young adults. By that age, mice that are born with CDKL5 deficiency already have all their symptoms, which include cognitive, motor and behavioral deficits, as well as alterations of neuronal excitability and synapses. Same as for people with CDD. And the first important finding of the publication is that if you let mice grow up to that age completely normal, and then turn off their CDKL5 gene, they will progressively develop almost all the same symptoms of the disease (except for less motor symptoms). Pretty much as if they had been born with CDKL5 deficiency.
And that is the first conclusion: CDKL5 deficiency looks like a disease of neuronal maintenance, not so much of neuronal development. We talked about that at the 2020 CDKL5 Forum (read it here).
The second part of the study gets better: if CDKL5 is so important for the mature brain, what happens if we bring it back at that point? How much can we get back?
For this the Penn team uses the second type of mice, the ones in which they can stop CDKL5 deficiency at any age they want. And they let them grow through all those key developmental ages with CDKL5 deficiency, and they let them display all of the disease symptoms. And then, when mice are early adults, they turned on the gene so they no longer lacked CDKL5. This is the mouse version of what would happen if we already had the perfect gene therapy for CDD and we treated patients that are no longer children.
And the results are beautiful and make this study a landmark in the development of curative treatments for CDD. Barbara Terzic and Joe and their colleagues show how later-age restoration of CDKL5 expression rescues most CDD deficits. They re-start CDKL5 expression in mice that are of young adult age, that already have developed all their CDD symptoms, and then see reduction or full rescue in many motor, cognitive and behavioral readouts. Mice with CDD don’t develop easy-to-measure seizures, which is a difference with how the disease presents in people, but the Penn group was able to interrogate brain circuit hyperexcitability using an excitatory neurotransmitter similar to glutamate to “poke” neurons, and saw how the hyperexcitability that comes with CDKL5 deficiency was also rescued by reintroduction of CDKL5 expression. They also used this neurotransmitter to induce seizures in the CDKL5 deficient mice since they are prone to epilepsy, and saw how this was also rescued by reintroduction of CDKL5 expression. They even looked at the presence of receptors in synapses, which is altered by CDKL5 deficiency, and this was also restored in the adult-rescued mice. So I want to highlight again how thorough and well-done this study is: it is very clear from this study that reintroducing CDKL5 expression later in life is able to correct functional and behavioral deficits across the board, not just a few symptoms.
And that is the big beautiful conclusion of the study: the promise of disease reversal in CDD, across a broader-than-expected time window.
WAS THIS NOT OBVIOUS OR KNOWN BEFORE?
No it wasn’t.

During brain development neurons need to be born, then need to migrate to where they are needed in the brain, grow connections to the parts of the brain that they need to be connected to, and then do their job properly remaining in place until we get old and neurons start dying. In many neurodevelopmental diseases neurons might fail to develop properly, or might migrate to the wrong places, or not connect with who they should, or suffer from neuronal loss. And for all those it is too late to come with the gene after those problems have happened.
We knew that CDD looked quite good for a potential genetic rescue in older individuals. We knew the neurons where there, in the right location, that brain connections looked good, and there were no signs of neurodegeneration. At the biological level CDD looks more like a channelopathy, like Dravet syndrome (Nav1.1 channel), than a proper developmental disease.
But from thinking “the odds are good” to being able to say “at least in mice this disease is reversible” there is a long stretch. That is why this study is such a landmark.
The results from the Adrian Bird study in Rett syndrome are actually an exception. Similar studies in related diseases have failed to show such an across-the-board rescue. For example in Phelan-McDermid mice, reintroduction of the SHANK3 gene in adult mice only rescues some symptoms, and early postnatal intervention is needed for across-the-board rescue (link). And in the Angelman mice (Ube3a gene), you can only rescue many of the symptoms if you restore the gene during early development, and the authors concluded that “adult reactivation of Ube3a is only minimally efficacious as a therapeutic intervention” (link).
So with CDD starting so early in patients, and with the clinical symptoms being so severe, it was not obvious at all that CDD would look so promising for rescue in older individuals and not more similar to the mouse study for Angelman.
WHAT IT MEANS FOR GENE THERAPIES
This study means that we now know that CDKL5 deficiency is biologically reversible. Now we need to get the tools to do that in the clinic.
What these genetic rescue studies in mice have in common is that they use mice that are genetically re-programmable, where we as scientists can choose to turn on or off a particular gene defect. People is not born like that, we cannot choose to turn off their genetic disease. We need therapies for that. These could be therapies like gene therapy, where we bring a new copy of the CDKL5 gene to their neurons, or other therapies like gene editing that fixes the mutation that they have, or even protein therapies if we can supply enough CDKL5 protein to their brain.
So there are many technical possibilities to restore CDKL5 in patients, but there are also many challenges to do this. This is why developing this type of treatment takes so long. One of the main challenges is getting to all (or most) neurons in the brain. CDD is not a disorder that impacts a small brain region, and where we only need to fix it there with an injection. It actually involves the entire brain, and we would need to make sure all (or many) of those neurons have sufficient CDKL5 as a result of a treatment. This sounds simple but is tremendously difficult. Understanding how many neurons need to be rescued to have enough efficacy, and how much CDKL5 exactly do they need (is it ok with half of the amount?), and having the right therapy that can do that in a safe way… it is not easy.
But the good news is that this is a challenge for ALL neurological diseases, so scientists and companies around the world are trying to make sure we know how to make treatments that can put back a gene (or fix it) throughout the brain. This will benefit CDD and many others. And it is also good news that we already have several of these scientists and companies working on developing these treatments for CDD, and we have already seen some early results with two of these gene therapies in mice with CDD (see here).
It will only get better from here. I expect this study to attract more attention, more research and more resources, to developing curative treatments for CDD now that we know biology is on our side.
In summary:
The part of the puzzle that we could do nothing about, whether the brain has a narrow temporal window where it needs CDKL5 or not, is the one where we got very lucky. CDKL5 deficiency is biologically reversible.
The second part of the puzzle is to build those therapies, and that one is not about luck, but about science and time and money. We can do much about it.
This is like the difficulty of building a rocket to go to the moon: it is hard, but you know that your destination is waiting at the other side.
Now that we know that moon exists, that the disease is biologically reversible, all hands on deck to build that rocket.
PS1: Thank you to the Penn teams for working so many years in this study. This study had financial support from the Loulou Foundation, IFCR and several NIH grants. Science is not easy or cheap. And thank you for making the publication open access!
PS2: The 2021 edition of the CDKL5 Forum is coming up in November 1-2, and this study is only one of the several good news that will be presented at the meeting.
Ana Mingorance, PhD
2021 CDKL5 ALLIANCE Virtual Family Conference
Last weekend, the CDKL5 deficiency disorder (CDD) community gathered in front of their computers for a virtual annual meeting organized by the International CDKL5 Alliance. For the 2021 Alliance meeting, the organizing team commission a series of pre-recorded videos to a large range of speakers and published the videos with subtitles so that all CDD families from all around the world could watch them at the same time. You can also access the videos at the CDKL5 Alliance website. Here is a personal summary of the 2021 CDKL5 Alliance meeting.
I miss conferences in person a lot. And the type of conference in person that I miss the most is the annual patient conferences organized by the rare epilepsy patient organizations that I collaborate with.
Last weekend, the CDKL5 deficiency disorder (CDD) community gathered in front of their computers for a virtual annual meeting organized by the International CDKL5 Alliance. Did you know that there are about 20 countries in the Alliance? That is how large and connected this community is. The last face-to-face meeting took place two years ago and I strongly encourage you to read the summary from that meeting.
For the 2021 Alliance meeting, the organizing team commissioned a series of pre-recorded videos to a large range of speakers and then published the videos with subtitles so that all CDD families from all around the world could watch them at the same time. You can also access the videos at the CDKL5 Alliance website.
Some parents of children with CDD have asked me to write a summary of the virtual conference, since a written summary is always easier to translate and digest. For all of you here is a personal summary of the 2021 CDKL5 Alliance meeting, sprinkled with some opinions as I often do.
1. THE SCIENCE: HIGHWAYS AND HEADQUARTERS
Prof John Rouse from Dundee reviewed where we are in our understanding of what CDKL5 does. In the recent years we have made large progresses on our understanding of what CDKL5 does in the cell and therefore what goes wrong when CDKL5 is missing.
Today we know that the protein CDKL5 turns other proteins on and off as if they had a light switch, and many of these proteins that are controlled by CDKL5 are associated with the neuronal skeleton (the cytoskeleton). The neuronal skeleton is not like our bones, which are essentially there to give us some structure. In cells, the skeleton is more like highways that are used to transport things to where they need to be, for example making sure that glutamate receptors get to the right spot in the surface of the neuron for the neuron-to-neuron connection to work fine.

And in addition to being important for the highways, it turns out that CDKL5 has a second job at the cell nucleus, which is basically the headquarters or control center in the cell from where all decisions are made. But what could CDK5 be doing there? This is what keeps John awake at night, so he has put together a team of scientists to try to answer this question. It turns out that in the nucleus, where all of the copies of all of our genes are located, the genes (made up of DNA) are constantly suffering small damages that the cell quickly repairs so that all genes work properly. And CDKL5 is a member of that DNA damage surveillance team. It does that by turning on other proteins that are needed to do the repair. And it also helps the cell read some of these genes. So CDKL5 is really important for the nucleus to function properly.
With the very complex (and multiple!) roles of CDKL5 in the cell, it is clear to me that the best way to fix the disease is to put the gene or the protein back, or to fix the mutation. Anything that will produce functional CDKL5 protein again. Trying to bypass a protein that is missing is doable if the function was only one, but for CDKL5 there are too many functions to try to fix, so I would focus on bringing CDKL5 back.
2. CLINICAL TRIALS: WHERE ARE WE TODAY
Before a drug or a gene therapy can be approved to be used to treat patients it has to progress through clinical trials. I think this year with the coronavirus vaccines we are all very familiar with how clinical trials work, so I will only explain that in rare diseases like CDD we have two stages of clinical trials: Phase 2 trials, where a small group of patients get the treatment, and Phase 3 trials, where many more patients from many countries get the treatment and usually half of them get placebo (but are offered the treatment at the end of the study anyways). If the Phase 3 is successful, then the company can apply for marketing authorisation. The entire process for trials and regulatory review for approval ends up taking 4 to 5 years.
We still don’t have gene or protein therapies for CDD, so while we wait for scientists to develop them we are trying to discover medicines that could treat the symptoms of CDD, helping all patients have a better life until we get to the curative treatments. We have had some clinical trials already with symptomatic treatments in CDD.
Last year we had very good news about clinical trials: the first Phase 3 trial in CDD, ganaxolone from Marinus, had been successful.
This year we also have very good news about clinical trials: we are about to start our second ever Phase 3 clinical trial in CDD, this time the drug is fenfluramine from Zogenix.
At the Alliance conference, Marinus reviewed their good data with ganaxolone in CDD. They are offering ganaxolone to all of the trial participants that want to keep taking it, and in the USA they are also offering it to other families who want to try ganaxolone before it gets approved. This is done through an Expanded Access Program. Ganaxolone works by acting on GABA, the inhibitory signal in the brain. If everything goes well ganaxolone will be the first drug to be approved for CDD.
Last year there were two Phase 2 studies in CDD. One was with the drug soticlestat, from Ovid and Takeda. This drug works by acting on the glutamate system, which is the excitatory signal in the brain. The companies were testing the drug in four different epilepsy syndromes, and after reviewing their data Takeda has announced that they will run Phase 3 studies in Dravet syndrome and Lennox-Gastaut syndrome. They still have not announced what they will do with CDD, and they did not present at the conference so we will have to keep an eye on any news about this program.
The second Phase 2 study from last year was with fenfluramine, which works by acting on the serotonin system, a modulatory signal in the brain. Fenfluramine has already been approved in the US and Europe for the treatment of Dravet syndrome, where it has impressive efficacy to reduce seizure frequency. Zogenix presented an overall introduction to the company at the Alliance meeting and didn’t talk much about their plans for CDD, but they have already announced that they are starting a Phase 3 trial in CDD with fenfluramine starting this year. Phase 3 trials are international, happening in many countries, so I look forward to the announcement of which countries will be included in this study. And if the Phase 3 results look like the small Phase 2 study results, we could have potentially a second drug approved for the symptomatic management of CDD.
This progression of clinical trials for CDD is exceptional! Only 5% of rare diseases have any drug approved, which means that 95% have nothing. And we are having these two Phase 3 trials back to back in CDD, so we are on track to join that 5%!
But these symptomatic treatments are only the tip of the iceberg. Right below we have a growing pipeline of therapies in development that will correct the lack of CDKL5.

3. THE FUTURE: FROM SYMPTOMS TO CURES
If you have a child with CDD the main answer that you probably wanted to get from this conference is WHEN are gene therapies starting clinical trials.
And I am sorry to say that the answer is “we don’t know yet”. Because we cannot know yet, nobody does. There were four different groups that presented data at the Alliance meeting showing their progresses towards developing a gene therapy for CDD. That is four different shots on goal, four independent efforts like when many companies all started developing vaccines for COVID without knowing how many would succeed and how fast it would happen. And we are following the same approach in CDD: many efforts all at the same time going as fast as they can.
The university of Bologna is developing a mix between an enzyme replacement therapy and a gene therapy. An enzyme replacement therapy is “protein therapy”, a type of treatment where you add the brain the protein that is missing, in our case the protein CDKL5 already made. For a gene therapy scientists use a virus to bring to cells a copy of the CDKL5. It is similar to the AstraZeneca or the Janssen vaccine but instead of using a virus to carry to the body the sequence of the coronavirus spike we use them to carry the sequence for the CDKL5 gene. The university of Bologna is using a virus to deliver to neurons the sequence for the CDKL5 gene, but it has been tweaked so that the protein CDKL5 will then be secreted by those neurons to all the surrounding ones, doing “local enzyme replacement therapy” on top of gene therapy. This is also called “cross-correction”. It is a cool idea. Amicus is doing something very very similar, in addition to classical enzyme replacement therapy with the purified protein. And then Ultragenyx and the University of Pennsylvania are doing the classical gene therapy using a virus to carry the sequence for the CDKL5 gene to the neurons in CDD. That means that we got to hear about FIVE different gene therapy or enzyme replacement approaches at the patient conference, by four groups since Amicus has two different programs. That is a lot.
The university of Bologna showed data using their cross-correction therapy in laboratory cells and mice. This is still early work at a university so it is too early to know how much work the experimental treatment still needs before it could move to trials.
Amicus presented some data about their cross-correction therapy program and their classical enzyme replacement therapy program. It was not an in-depth project review, but more of an overview of all the work that Amicus is doing in CDD which includes not only these two programs but also helping understand better how CDKL5 works and developing biomarkers (detection of special signals using EEG) that will help check if therapies are working in CDD mice and patients much faster. CDD is clearly important to Amicus. They are doing many experiments in mice with both treatments to see how well they could prevent or reverse the symptom of the disease in mice with CDKL5 deficiency.
The University of Pennsylvania presented a lot of data in mice with CDKL5 deficiency using a gene therapy to show that it is possible to prevent most of the symptoms in CDD mice. I say “prevent” because they give the gene therapy to mice when they are babies. I really liked the presentation, because they showed how the gene therapy was effective in many disease symptoms. That is the main different with symptomatic treatments: with gene therapy, you put the gene back and you are treating all of the different symptoms at once by removing the cause of all of them. This group has also run initial experiments in larger animals (macaque monkeys) to check safety and because we need to check that the gene therapy can get to enough neurons in larger brains, which is literally a much larger challenge than delivering the gene therapy to the brain of a mouse which is the size of your thumb nail. They explained that they are currently optimizing the gene therapy so that then they can go to trials.
The last presentation was from Ultragenyx, who are also developing a classical gene therapy similar to the one from the University of Pennsylvania. I also liked this presentation very much because they treated CDD mice at older ages, the equivalent of children 4-11 years of age, so what they showed to us was that it is possible to reverse many of the symptoms in CDD mice. They saw reduction of cognitive, motor and behavioral symptoms in the mice, and even used the gene therapy to correct neuronal connection problems in “human neurons” which were produced from patient skin biopsies and turned into neurons in the lab. Just like for the University of Pennsylvania, they explained that they are currently optimizing the gene therapy to be able to get into many neurons in larger brains (monkey) so that then they can go to trials.
As soon as one of the groups considers that their gene therapy is optimized enough, they will start the process to request a clinical trial permission and at that point we will finally get a date for when clinical trials might start. I hope you all realize how much work all of this is, and how lucky we are to have these five protein and gene therapy programs being all developed at the same time for CDD!
4. DATA, DATA, DATA
There were several presentations about why the international CDKL5 patient registry is so important, the international CDKL5 patient database from Australia, a study to validate several severity and symptom assessment for CDD, and an international observational study that is about to start and that is coordinated by us at the Loulou Foundation.
At the core of all of these efforts is a common theme: we need to understand better the symptoms of CDD and how they change over time (the natural history), we need to identify good scales to measure these symptoms, and this information and scales are absolutely essential to be able to tell if a treatment is working beyond just reducing seizures.
For the registry and the database, you can provide data about your child from your living room, and for the observational study we will try to get as close to your town and living room as possible!
By the way, have you noticed that clinical trials all have names? For example the trial with ganaxolone in CDD is the Marigold study, and the famous trials with spinraza in SMA was called Nurture. The Loulou Foundation is requesting suggestions from the community to name the international observational study so please reach out with your proposed names! (you can get some ideas here)
5. THE COMMUNITY
Although we didn’t get to meet in person, we did get to see many of the visible faces of the CDD patient community at this virtual conference.
Patric Benz from the Swiss CDKL5 patient group was the meeting host on behalf of the German-speaking national patient associations who were the organizers. His greeting in several languages and the fact that the videos were available with multiple language subtitles was a testimony to the reality of rare disease patient communities: at the same time dispersed and united.
During the conference we also saw images of people with CDD kayaking, skiing, ice skating and cycling. If there is something notable about this community it also their fearlessness and determination.
To conclude the conference, Nathalie Ladly from CDKL5 Canada and Antonino Caridi from CDKL5 Italy spoke about some of the big efforts of the Alliance in this last year, like developing the organization manifesto and sourcing a list of all possible clinical trial sites world-wide recommended by the community so that we can accelerate clinical trials. The goal of the Alliance is to expedite treatments and a cure for CDKL5, so of course a global pandemic would not stop them from putting together such a great conference program.
Next stop is the CDKL5 Forum in November, also full of updates about the treatment programs in development for CDD. Hopefully by then we get to meet in person.
I hope you enjoyed this summary! let me know your thoughts in the comments.
Ana Mingorance, PhD
Disclaimer: These are my own impressions from the presentations that I was most interested in, and not an official text about the Alliance meeting by Alliance or the Loulou Foundation. I write these texts with the parents of people with CDD in mind, so excuse also my lack of technical accuracy in parts ;-)

Dravet syndrome pipeline review 2020 now available
As of end 2020, the Dravet syndrome pipeline comprises 3 approved drugs, 11 drug candidates, and 12 different products have received orphan drug designations. Since the last report the pipeline has changed by, among others, the approval by FDA and EMA of Epidyolex (cannabidiol) and Fintepla (fenfluramine), the initiation of the first clinical trial with an antisense therapy, and progresses in different disease-targeting modalities including gene therapy approaches.
Dravet syndrome pipeline and opportunities review 2020 is a market research publication that provides an overview of the global therapeutic landscape of Dravet syndrome, an orphan epilepsy disorder with multiple non-seizure comorbidities and high unmet medical need.
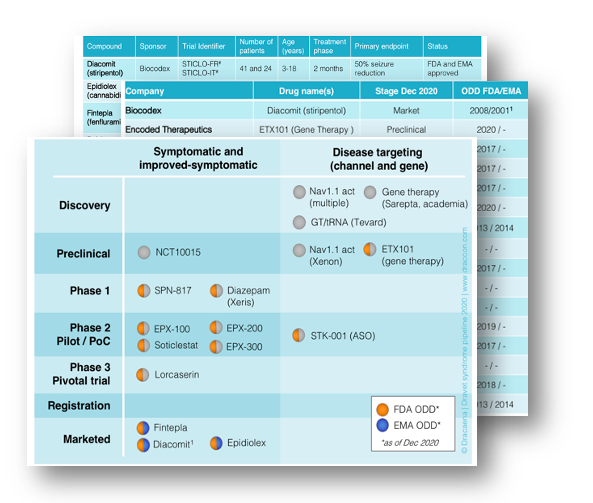
As of end 2020, the Dravet syndrome pipeline comprises 3 approved drugs, 11 drug candidates, and 12 different products have received orphan drug designations. Since the last report the pipeline has changed by, among others, the approval by FDA and EMA of Epidyolex (cannabidiol) and Fintepla (fenfluramine), the initiation of the first clinical trial with an antisense therapy, and progresses in different disease-targeting modalities including gene therapy approaches.
The 2020 pipeline report includes the most recent updates on programs from Biocodex (stiripentol), Encoded Therapeutics (ETX101), Eisai (lorcaserin), Epygenix Therapeutics (EPK-100, -200 and -300), GW Pharmaceuticals (Epidiolex / Epidyolex), INSYS Therapeutics, Lundbeck, NeuroCycle (NCT10015), OPKO Health (OPK88001, CUR-1915), Ovid Therapeutics (Soticlestat, OV935), PTC Therapeutics (ataluren), Sarepta, StrideBio, Stoke Therapeutics (STK-001), Supernus Pharmaceuticals (SPN-817, Huperzine), Takeda Pharmaceutical (Soticlestat, TAK-935), Tevard Biosciences, Xeris Pharmaceuticals (XP-0863), Zogenix (Fintepla, ZX008) and Zynerba Pharmaceuticals.
The Dravet syndrome pipeline and opportunities review 2020 report also includes an analysis of the competitive landscape, unmet needs, and evaluates current and future opportunities of the Dravet syndrome market.
You can find the 2020 pipeline review, as well as the 2019, 2018 and 2017 editions here:

Top 5 insights from the American Epilepsy Society meeting (2020)
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. This year the meeting was virtual, which made the exhibit hall and the poster sessions less exciting but also made the presentations more accessible, including a 90-day on-demand period after the meeting to catch up with all of the parallel tracks. These are my own 5 insights from the American Epilepsy Society 2020 meeting.
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. I look forward every year to the first week of December for this reason.
This year the meeting was virtual, which made the exhibit hall and the poster sessions less exciting but also made the presentations more accessible, including a 90-day on-demand period after the meeting to catch up with all of the parallel tracks.
I look for therapies for rare genetic epilepsies, and in the recent years this area has exploded to take over much of the AES meeting. Because of that I will focus this update on the rare genetic epilepsies, which come under many names like Developmental and Epileptic Encephalopathies (DEEs) since not all of them are genetic. There is a lot more that is presented at the AES meeting and that I will not cover. These are my own 5 insights from the American Epilepsy Society 2020 meeting.
1. DEVELOPMENTAL AND EPILEPTIC ENCEPHALOPATHIES AT THE CENTER OF THE AGENDA
The main difference between DEEs and general epilepsy is not their frequency. DEEs are largely genetic, and have many more impacts than just seizures, so as the DEEs take more space in the agenda at AES, the conversation also turns from seizures to non-seizure comborbidities, and from symptomatic management to genetic treatments.

This year the discussion around DEEs was again very prominent in the conference agenda, making it hard to miss by attending clinicians. For example, we opened the congress with a Presidential Symposium focused on DEES. There was also a very interesting Investigator Workshop dedicated to pediatric trials, particularly relevant for these patient populations, and another Investigator Workshop that I moderated dedicated to genetic treatments in DEEs. And we had different industry-sponsored symptosia around rare childhood epilepsies including DEEs as well.
Going through my notes on how the discussion around DEEs has evolved from one year to the next at AES we can track that increasing prominence:
In 2017, there was already growing attention for the “orphan epilepsies” and everybody was excited about cannabidiol and fenfluramine. It was starting to become clear that there are just too many genetic syndromes to try to diagnose them one by one, and that we need to run genetic epilepsy panels as soon as possible in children and reevaluate adults. AES 2017 was a great meeting, but I was still hoping that we could move beyond genetic diagnosis and towards genetic treatments.
Then we made a big jump in that direction at the American Epilepsy Society meeting in 2018. I wrote in that article: “There was one key progress visible at the AES 2018 meeting that defines a before and after moment in the field of epilepsy, and this is the arrival of non-pharmacological therapies for treating epilepsy.”
That “before and after” was even more clear during last year’s conference. During AES 2019, the sessions on genetic therapies and antisense oligonucleotide (ASO) therapies for rare epilepsies were filled to full (standing) capacity and the organizers had to close the doors. The epilepsy field, which has been until recently dominated by pharmacology, had embraced the technologies of biotech and we have officially entered a new era. I wrote that “AES 2019 was the year of genetic therapies for the developmental and epileptic encephalopathies”.
AES 2020 provided a continuation to these discussions, with greater visibility for the DEEs, their particular challenges and the treatments in development that target their biological cause. This prominence is turning the conversation from one of interest mainly for a subgroup of people (like me!) into a central topic in epilepsy community.
And this is very important, because as these conversations are featured more often at the congress and also in the major sessions, they reach a broader audience of practicing neurologists and epileptologists who might not often get exposed to the genetic epilepsies. My hope is that this will translate into more awareness, with more patients being evaluated for potential genetic syndromes and with that more diagnosis. And with more diagnosis we get more targeted therapies researched and taken into the clinic, so it is critical to get that first ball rolling: the one of broader awareness among clinicians.
2. BIG PROGRESS IN DRAVET SYNDROME
The poster child for how the epilepsy field is moving from treating mainly symptoms to chasing cures (genes), is Dravet syndrome. And that is because Dravet syndrome holds one foot in the classical clinical epilepsy syndromes (it is not a genetic diagnosis), and one foot into the mono-genetic disorder space, with a large majority of patients having SCN1A haploinsufficiency. So it was the ideal “transitional disorder”.
Because of that, we can see in Dravet syndrome an impressive progress in barely a decade from having no clinical trials, to having initial trials with symptomatic treatments, to having multiple symptomatic treatment approvals, to now having genetic therapies in development. I expect many other syndromes to follow, probably with faster timelines to genetic therapies.
2020 was a very good year for Dravet syndrome and that was seen in the AES 2020 meeting. This is a very-short summary of the progresses in Dravet syndrome therapy development presented at AES 2020:
The European approval of Fintepla (fenfluramine) for Dravet syndrome makes it the 3rd drug specifically approved for treatment seizures in Dravet syndrome, together with Diacomit (stiripentol) and Epidiolex/Epidyolex (cannabidiol). We could see data at the conference on fenfluramine’s long term efficacy in adult patients with Dravet syndrome as well as long-term safety data.
Takeda and Ovid Therapeutics presented data on a Phase 2 study with the drug Soticlestat, an indirect modulator of NMDA activity, with about 34% seizure reduction in the placebo-controlled study, similar to cannabidiol. The companies have announced their intent to pursue a Phase 3 program for Soticlestat in Dravet syndrome in the near future.
In preparation for clinical trials that can measure non-seizure outcomes in Dravet syndrome, Stoke is running an observational study called BUTTERFLY, and they presented at AES their first results indicating the feasibility of a small collection of clinical scales to measure these outcomes. This might not sound very exciting, but it is a milestone study that will help us transition from running clinical trials in Dravet syndrome as epilepsy trials, to running them as neurodevelopmental disorder trials – and those are the ones we need for genetic therapies.
Staying with Stoke, the company is developing an ASO to increase functional SCN1A mRNA and protein expression and dosed their first patient some months ago, making it the first patient with Dravet syndrome to ever receive a therapy designed to rescue their genetic problem, another major milestone study for the field. At AES, Stoke presented their Phase 1b/2a trial design (MONARCH study), that focuses on PK, safety and tolerability but will also produce some early efficacy signal. The company expects to have some early data from this study in 2021.
A bit earlier in the pipeline, Encoded Therapeutics is using an AAV approach for Dravet syndrome where they drive the expression of a transcriptional activator for SCN1A only in interneurons. The purpose is to increase SCN1A and therefore Nav1.1 only in interneurons, which are the main neuronal population that needs it. Last year they presented their preclinical proof-of-concept in mice and this year they showed they had taken a large step towards the clinic presenting the biodistribution and safety of their gene therapy in non-human primates. The company has announced that their one-time treatment for Dravet could start clinical trials in 2021.
There are additional gene therapies for Dravet syndrome in development, and as part of the Investigator Workshop that I organized, Dr Ricobaraza from CIMA presented an adenovirus-based experimental therapy that they are developing to supplement SCN1A expression in the brain. This therapy showed broad rescue of disease phenotypes in SCN1A mutant mice and uses a different approach to the gene therapy from Encoded by supplementing SCN1A expression instead of boosting endogenous expression.
And Xenon Pharma, a Canadian biotech company, presented early proof-of-principle in mice for a small molecule Nav1.1 activator. The discovery of this class of compounds was one of my favorite poster presentations from 2019 and this year Xenon was able to come with additional data, including in mice with SCN1A haploinsufficiency. Following an ASO and a viral-delivered gene therapy, a small molecule able to increase Nav1.1 activity can be a game changes and reminds me of risdipram, the small molecule from Roche for the treatment of SMN.
If your main interest is in this syndrome, I recommend you to also read the AES summary from the Dravet Syndrome Foundation.
3. AND NOT JUST DRAVET SYNDROME
The industry interest in orphan indications within the epilepsy space doesn’t stop at Dravet syndrome. The adoption of genetic panels for diagnosis of pediatric neurological conditions has led to the identification of many other syndromes that present with epilepsy, and many more patients with each of these conditions. This has helped us describe them as new disorders and start clinical trials looking for effective medications.
A disorder that is following the steps of Dravet syndrome is CDKL5 Deficiency Disorder (CDD).
At AES 2020 we saw the results of the first pivotal trial in CDD, a 101-patient Phase 3 trial with ganaxolone (by Marinus Pharmaceuticals). This clinical trial, called the Marigold study, demonstrated efficacy of ganaxolone in reducing seizures in this population and might lead to the first approval for CDD. The company plans to apply for marketing authorization in 2021. Something that caught my eye looking at the trial data is that at baseline, CDD patients have 50-60 major motor seizures per month, stressing both the severity of the disorder and the urgent need for effective medications.
Dr Devinsky from NYU presented the results of an ongoing open-label Phase 2 study with fenfluramine in CDD, with 6 patients with CDD showing a 90% reduction in tonic-clonic seizures. This makes fenfluramine a very promising potential treatment for CDD, and I hope to see it evaluated in larger placebo-controlled studies for this disorder.

Dr Devinsky also presented a review of all clinical trials so far in CDD, including clinical trial data with cannabidiol, ataluren and soticlestat. This is important because many therapeutic programs in development for CDD were not presented at the AES 2020 meeting, and if we hadn’t seen this review we would think that there are fewer developments in CDD.
This is one of my main observations from the meeting, which I explore further in the next point. We had several presentations on the different DEEs, often as a group, but a lot of the therapeutic developments were missing from the conference. In particular we were missing virtually all of the genetic treatments in early development.
We saw trial data in Lennox-Gastaut Syndrome with Soticlestat (Phase 2) and fenfluramine (Phase 3), and early clinical data on the pharmacokinetics of NBI-921352/XEN901, a Nav1.6 inhibitor, that is being developed for SCN8A developmental and epileptic encephalopathy and that I look forward to seeing it reach patients in future trials.
But there is so much more that was missing from the conference! I would have liked to know how the ASOs and small molecules for SCN2A-associated disorders are doing, how the KCNQ2 modulators from Xenon and Knopp are doing, if there are progresses with the ASO for KCNT1 epileptic encephalopathy, and if there are any new therapeutic developments to target SynGAP1 or STXBP1 or PCDH19 or many others. And that all leads me to my next point….
4. SOME OF THE BEST UPDATES OF THE YEAR WERE MISSING FROM AES
I know that many of my readers are parents of children with rare epilepsy syndromes, so I want to stress this message: there is a lot that was not presented at AES 2020. If there was nothing (or little) presented about your child’s disease it DOES NOT mean that there is no reach being done on treatments for their disease.
So while the AES congress is the best meeting of the year for epilepsy, it does not get deep enough in all of the different areas, in particular when it comes to academic efforts or some genetic approaches. This is not the Society’s fault. A small part of the program is chosen by AES but the large bulk of it is determined bottom-up: clinicians and scientists (and some times patient groups) submit proposals for sessions, such as investigator workshops, or for individual presentations, and these are then selected based on quality and interest in the area – and clearly AES has seen a major interest for DEEs and genetic treatments. But if people don’t submit some project for presentation at AES, it will be missing. And a lot is missing.
Where can you find those project updates? Quite a bit of it will be presented at antisense oligonucleotide meetings or gene therapy meetings while they are still in early development. For example, this year there were two different gene therapies for CDD that were presented at the American Society for Gene and Cell Therapy meeting, and you would have never known this had happened if you only attended AES 2020. Much of the academic work doesn’t get presented at “clinician” conferences either, and they are more likely to be presented at neuroscience meetings for example.
And hands down the best place where you will find all this information is at the scientific meetings organized by the patient community. These are the best meetings to get a complete view of all of what is coming up for each disorder. At the CDKL5 Forum organized by the Loulou Foundation we review every year the state of all research efforts and see presentations by the teams driving this science. Other examples are the Dravet Syndrome Foundation Biennial Conference, the SynGap Research Fund Roundtable, the Foundation for Angelman Syndrome Therapeutics (FAST) Global Summit and many others.
And this latest one is important to highlight, because the treatment development announcement that I consider the most groundbreaking one of 2020 is the early data in Angelman syndrome from GeneTx and Ultragenyx. Their treatment, GTX-102, is an ASO that allows for the unsilencing of the second copy of UBE3A in patients with Angelman syndrome. Preliminary data from the first 5 patients with Angelman syndrome treated with the ASO showed very large improvement in all of them with reports of efficacy that we simply didn’t know were possible in just 4 months (or at all) in a neurodevelopmental disease. Improvements like previously non-verbal patients acquiring spoken language, learning to respond to their name or learning to self-feed with a fork. All of this in 4 months! There are some safety concerns with this therapy that the companies hope to be able to manage, but what is most important to me is what we have just seen as biologically possible.

I hope all families of patients with DEEs have heard about these data. And this was a finding that will probably never make it to AES because it was not about seizures, but it is the holy grail that we look for in DEEs, which is why I stress the need to make sure we attend the meetings organized by the research foundations and patient organizations, so that we know the true state of scientific development in the space.
Coming back to AES, I am very happy with the general conversation featuring so prominently the DEEs, but not with how much (how little?) we get to hear about therapeutic programs in development for each of the DEEs. In addition to following the developments in those disorders through the separate disease-focused meetings, I encourage us all to submit proposals about these disorders to be presented at AES 2021, and perhaps we can try to include more presentations that review the pipeline for the syndromes like the one from Dr Devinsky for CDD. That will help us get a more complete overview of treatments in development at AES.
5. DIFFERENT TRIALS, DIFFERENT CHALLENGES
A very interesting conversation at AES is that the traditional clinical trial design in epilepsy will not be enough for all the new syndromes or for all the new treatments.

We talked about the challenge with “ultra-rare” diseases, where you might have few diagnosed with that disease, or perhaps a treatment (like a mutation-targeted ASO) that could only work in a handful of patients at most. In these cases, it is too hard to satisfy the usual regulatory requirements for running a trial, so some patients and some companies are pushing for n-of-1 “trials”. And I use quotation marks around the word trials in this case, because as Dr Adam Numis from UCSF puts so well into words: “is this research or is this clinical care?”. And if we are essentially using those trials for clinical care, we need to be cognizant that they don’t represent proper research so they might end up not being reproducible (like quinidine and KCNT1).
We also talked about non-seizure outcome measures and endpoints for clinical trials, which we all want to see in the DEEs. Some observational studies are starting to identify some possible outcome measures, but then we hit the next challenge: how long do trials need to be to see an improvement in some of these outcomes? And would it be ethical to have patients wait in a placebo group all that time? I really enjoyed Dr Madison Berl’s presentation about some non-seizure outcomes that could potentially show a change earlier, and her recommendation to use raw scores in these clinical scales (not the total scores) to focus on items that are more specific for that patient population and likely to respond sooner.
About this, a related discussion that I would like to hear more about next year is how we will interpret all of those non-seizure outcomes and where to trace the line for something being clinically meaningful. In the patient community they speak about “inchstones”, the little accomplishments that might not usually count as a developmental milestone but that really make a big different to patients and their families. My hope is that genetic treatments will be so impressive that it will be obvious that they improve patient outcomes, like in the SMA trials, but until we have those treatments we need to identify the best way to capture these inchstones as well.
LOOKING INTO 2021
I think we are all happy to get done with 2020… for many reasons. As we look into the new year there are some planned milestones that I look forward to, and some things that I hope will also come true:
I look forward to Stoke early results in Dravet syndrome with STK001 (their ASO for SCN1A) and the initiation of clinical trials with ETX101 (gene therapy) from Encoded Therapeutics.
I look forward to ganaxolone filing for approval for CDD.
I look forward to more phase 3 studies in more DEEs, including Soticlestat and fenfluramine and potentially others.
I look forward to more ASOs and gene therapies approaching clinic. I want to see more proof-of-concept in mice, biodistribution studies, and IND-enabling programs. Step-by-step getting us close to trials.
I also keep an eye on those small molecules that also target the disease and that could be transformational for some channelopathies.
I look forward to many research foundation and patient organization-led conferences in 2021, virtual or not, where we can get a view of all efforts ongoing on these disorders and establish collaborations and networks.
And I look forward to more beautiful surprises, like the ASO results in Angelman, that remind us that we still don’t know what biology can do if we come up with the right therapy.
Ana Mingorance, PhD

MAIN LESSONS FROM THE 2020 CDKL5 FORUM
For the past six years the Loulou Foundation hosts an annual meeting where scientists and drug developers working on CDKL5 deficiency, together with representatives from patient organizations, meet to discuss the latest advances. This was the fourth Forum I attended, and my third since joining the Loulou Foundation.
Here are the main news and take-home messages from the 2020 CDKL5 Forum that took place in October 12-14 2020.
Artículo en Español en este enlace
2020 is the year when the world discovered the urgency that the rare disease community feels to develop effective medications and cures.
It is also the year that prevented us from hosting large conferences, so the 6th edition of the CDKL5 Forum, which would have taken place in London, UK, instead took place October 12-14 in a virtual conference center. The Forum is the annual meeting hosted by the Loulou Foundation where scientists and drug developers working on CDKL5 Deficiency Disorder (CDD), together with representatives from patient organizations, meet to discuss the latest advances. This was the fourth Forum I attended, and the third since joining the Loulou Foundation.

Every year, the CDKL5 Forum allows us to look back in time to how the field was just a couple of years ago, review all the progresses made in the last year, and anticipate how the next year of research into CDD and drug development is likely to look like. And if we are to learn from our predictions from the past years, we will probably underestimate how much everything will move in just 12 months.
Dr Orrin Devinsky, Chairman of the Loulou Foundation Advisory Committee, summarized the 2020 Forum stating how “what we have seen these 3 days is breathtaking”. For those of you who were not in the room, these are my main conclusions from the CDKL5 Forum and how far along we have come in the CDD field.
1. UNEXPECTED WONDERFUL BIOLOGY
The Forum always starts by updating our growing understanding of what the CDKL5 protein does and why mutations in the CDKL5 gene lead to such a dramatic neurodevelopmental condition.
I am a cell biologist and I am used to thinking about genetic conditions by knowing what the missing (or mutated) protein does. For example, “this is an ion channel so when it is missing neurons are hyperexcitable”, or “this protein is essential for protein degradation so when it is mutated there is abnormal buildup of toxic proteins”. But CDKL5 deficiency is challenging all those simple schemes. The CDKL5 protein is a kinase involved in so many cellular processes that learning about what happens when CDKL5 is missing is not only teaching us about how the deficiency disorder works, it is also teaching us new biology previously unknown to science.
In the words of Dr Victor Faundez from Emory University, which capture very well this take-home message:
““One mutant opened the door to wonderful biology””
We saw much of this during a pre-meeting with the Loulou Foundation awardees as well as during the first day of the Forum. Scientist from all around the globe shared their findings about how CDKL5 modulates microtubule dynamics (like the skeleton within cells), and how this is so critical for neuronal plasticity and formation of new synapses (neuronal connections). And CDKL5 also seems to have important functions at the cellular headquarter where DNA is located: the nucleus. They also told us how CDKL5 also regulates motile cilia, like in the epithelial cells that surround the brain ventricles and move CSF around, and about functions outside of the brain. And CDKL5 starts looking like a kinase that probably controls many other kinases, like a critical node in a network, so it takes us in many directions. From some of these pathways we start identifying “actionable” targets, which suggest possible drugs that might be used to at last ameliorate some of these consequences of missing CDKL5.
That said, it is clear that with CDKL5 controlling so many processes it will be hard to “bypass it” or achieve a full rescue with treatments that target individual pathways. We really should try to replace the protein or the gene to achieve the full recovery. The good news is that these treatments are all in development.
Some of the consequences of missing CDKL5 remind me to what goes wrong in other neurodevelopmental disorders. For example, all of the microtubule dynamic discussions make me think of KIF1A associated neurological disorder, and a recent publication showing that CDKL5 could be flagging proteins for ubiquitination in the kidney reminds me to Angelman syndrome, where a missing ubiquitin ligase is the culprit. So these all make sense as individual pathways that once de-regulated by the lack of CDKL5 might lead to the neurodevelopmental disorder, and they happen to all be engaged at the same time in CDD.
All because of the one mutant that opened the door to wonderful biology.
2. PACKING THE TRANSLATIONAL TOOLBOX
One of the reasons why so many scientists have come to work on CDKL5 is the biology that it uncovers. Another reason is research tools.

Scientist cannot research a protein or a disease if they don’t have the tools to produce that protein, or to see its activity, or to model the disease in cells or animals. A big focus of the Loulou Foundation and our partner organizations has been to build “the translational toolbox” so that we have all we need to take a therapy all the way from understanding what happens in the cell to completing clinical trials. Last year the Forum dedicated the parallel breakout sessions to map out the needs for tools and to focus our efforts to create them. A year later, those efforts have paid off.
I will only highlight three of those tools, there were more presented:
Dan Lavery, CSO of the Loulou Foundation and our Forum host, announced that Coriell (a biomedical tool repository) will soon start offering iPSCs from CDD patients. This is the result of years of work in collaboration with the Boston Children’s Hospital that led to the creation of several lines of iPSCs together with isogenic controls. For the non-scientists: iPSCs are inducible pluripotent stem cells, a sort of stem cell that is obtained by taking some cells from the blood or the skin of a person (in this case CDD patients) and “reverting” them to stem cell state. They are very useful because scientists can then make them go forward in development, this time to neurons, and be able to have CDD neurons in a dish. And because they will be available from this repository, getting them will soon be as easy for scientists as placing an order on Amazon.
Another important tool to research CDKL5 is CDKL5 itself. Brian Ranes from Amicus Therapeutics offered to share purified CDKL5 protein with the research community, opening the door to exciting science that was until now very difficult to do because producing and purifying CDKL5 is remarkably complicated. But Amicus has developed considerable expertise around protein production, and showed once again their commitment to advancing research in CDD beyond just working on their own therapeutic programs.
One critical challenge in the development of therapies is reproducibility, in particular reproducibility of the preclinical (mouse) drug studies. A year ago, the Loulou Foundation signed a joint collaboration with Baylor College of Medicine and the Texas Children’s Hospital to establish a pipeline of CDD mouse models and behavioral assays to be able to evaluate (and confirm) potential therapeutics for CDD in a rigorous and unbiased way. The work is led by Dr Rodney Samaco, who was the recipient of the Lab of the Year 2020 award by the Loulou Foundation for the transformational value that this mouse behavioral assay platform will have for the development of therapeutics in CDD. This platform has already been instrumental for the preclinical proof-of-concept for the gene therapy from Ultragenyx that was also presented at the Forum (more information below).
Building the research toolbox takes many years, many scientists, much funding and much collaboration. The CDD translational toolbox is a reflection of the maturity of the CDD field.
3. CDD IS TECHNICALLY NOT A NEURODEVELOPMENTAL DISORDER
This provocative statement is one of the major conclusions that I take from the 2020 CDKL5 Forum.
Coming into this Forum we already knew that there are no large changes in neuronal wiring in CDD, or in brain anatomy, and we have not seen signs of it being a neurodegenerative disease. So it would appear that CDKL5 is constantly needed for the very dynamic process of synaptic plasticity and formation. This means that bringing CDKL5 expression back is likely to provide a benefit even in the more mature brains, while in diseases that affect neuronal migration, or that lead to neuronal death, we only have a narrow time window to replace the protein and see any improvement. But we really had no evidence for this yet.
During this Forum we learned from mouse studies that the CDKL5 protein remains highly expressed throughout life (so it is not only there during early development). We also learnt that if we remove it from the brain of an adult mouse that has otherwise developed completely normal, as if CDD could be started in an adult individual by flipping a switch, this results in all of the same problems that mice have if they are born with CDD. Joe Zhou from the University of Pennsylvania showed us how removal of CDKL5 expression in adult mice resulted in hyperactivity, reduced sociability and other autism-spectrum behaviors, memory problems and poor motor coordination (this is how CDD looks in mice, remember that it is very hard and rare to see seizures in CDD mice). Because removal of CDKL5 at a later stage is enough to produce the disease, Joe’s conclusion was that “CDKL5 is indispensable for life”.
But what about the reverse? What is the consequence of bringing CDKL5 expression back to a brain that has developed without it? Joe told us how he plans to do the reverse mouse, one where the animals are born without CDKL5 and then he can start the gene at different ages, and we all look forward to seeing those results. In the meantime, the first partial answers to these questions came from the gene therapy programs. Sharyl Fyffe-Maricich from Ultragenyx showed how administering AAV carrying a copy of CDKL5 to mice with CDD aged 3 to 5 weeks, which is the equivalent of children 4 to 11 years old, was able to improve several of the phenotypes of these mice, including motor function and cognition. While she made it clear that they were only able to restore CDKL5 expression to 35% of its usual levels, which is just a partial rescue, these experiments showed that at least in mice even partial rescue of CDKL5 expression is sufficient to reduce the disease symptoms, and this is the first time that we have been able to measure this directly.
What this tells us is that CDD is not strictly a neurodevelopmental disorder, where the CDKL5 protein is needed for brain development and no longer needed afterwards. Instead, CDD is a neuromaintenance disorder, where CDKL5 protein expression is needed throughout life to ensure proper neuronal functioning. We don’t appear to be fighting against a transient neurodevelopmental window when CDKL5 is needed and where the damage is already done if we come too late. And this is much more hopeful for treatments, because all of these experiments tell us that CDD is a good candidate for gene therapy or enzyme replacement therapy even in adult patients.
“CDD is not a neurodevelopmental disorder, but a neuromaintenance disorder, and this is much more hopeful for treatments.
”
4. GENE THERAPIES (IN PLURAL) ARE COMING
The topic of gene therapies was a major one in day 2 in the 2020 Forum. Jim Wilson, a veteran from the gene therapy field, explained us how gene therapies have to progress roughly through 3 stages before they can be used to treat large groups of patients in clinical trials. I will give them a name based on my own words:
- Stage 1 – Prove that you can treat a mouse: the first treatment studies are often done in mice that carry the same genetic deficit that the patients have, to test the ability to correct the disorder in these animals with the experimental therapy and get an early feeling of potential toxicity issues. In gene therapy scientists use a virus that they have emptied from virus DNA and replaced by the therapeutic gene that is needed for the patient, so that when the virus infects the patient it is actually bringing the gene to the cells. So the biggest challenge of this stage is to produce that “transgenic therapeutic virus” which involves several pieces, and to treat the CDD mice with it.
- Stage 2 – Prove that you can treat larger brains: these are the toxicology studies which are done in large animals, often non-human primates, looking for safety and ability to bring the virus to large parts of the brain. This is often the biggest challenge, the “biodistribution” of the virus, in particular for diseases that need to treat large areas of the brain (or other organs) as opposed to very focal diseases.
- Stage 3 – First in Human studies: this is when the therapy reaches clinic for the first time, and the first studies focus on safety and toxicology before risking exposing too many patients to the new experimental gene therapy.
Jim reviewed how the challenge for many gene therapies is step 2, and figuring out how to scale up from very small mice to large animals. For example, figuring out the route of administration. In the case of CDD, could we use intravenous administration or should we deliver the virus directly into the brain? Earlier in the program we had learned from Amicus that they are working on a project to boost the number of neurons that can benefit from gene therapy by using a virus to deliver not the plain CDKL5 gene but a gene encoding for a secretable CDKL5, so that the neurons that receive the virus and can express CDKL5 will also act as local factories to secrete CDKL5 that can then get into neighboring cells, an approach that they called “cross-correction”.
And then we got to one of the most exciting moments in the Forum, when we got to see two proof-of-concept in mice from two companies developing gene therapies for CDD.
The first one was from Jim Wilson’s group, who is running a gene therapy for CDD program for a company called Elaaj Bio. Ralf Smichd presented a very large collection of experiments optimizing the gene therapy for CDD. He showed us how much work went into creating the “transgenic therapeutic virus”, from seeing which of the different isoforms of CDKL5 is needed (there are 4!) to checking what happens if you overexpress CDKL5 in the brain (to know if we face an overexpression risk) to confirming activity in both males and females. And to run the proof-of-concept, this group chose to go for the best-case scenario, which is to treat CDD mice at birth. This ensures the greatest brain coverage for the virus as well as treating as early as possible. The best-case scenario in CDD mice was amazing. Ralf showed complete rescue of CDKL5 protein expression AND activity, because we can monitor the kinase activity looking at a reporter phospho-target called EB2. He showed us beautiful brain expression of CDKL5, and rescue of all the symptoms in these mice when examined months after the virus administration into their CSF. He even showed some preliminary EEG improvement, and had reproduced these data in a separate lab with two additional CDD mouse strains. You can hardly get more solid science than here.
This gene therapy is now being optimized for the stage 2 that Jim explained to us before, which is to tweak it until it has good biodistribution in the non-human primate (large) brain so that they can confirm toxicity and safety and best dosing in these large animals before thinking about trials.
And as I commented before, Ultragenyx also presented a mouse proof-of-concept with their own gene therapy, which earned them the 2020 CDKL5 Forum Award for the “Company making a difference” in the preclinical space. And I found very interesting that Ultragenix had chosen a different approach for designing their mouse proof-of-concept. Instead of going for the best-case scenario, as in the previous therapy, Ultragenyx chose to go for the real-life-scenario, which was to treat older mice with a partial rescue of gene expression. And Sharyl showed us how even partial rescue with the virus administered directly into the CSF of CDD mice at the equivalent human age of roughly 4 to 11 years old resulted in partial efficacy across different symptoms. The results were therefore tremendously encouraging because we are likely to achieve only partial rescue in the clinic and we will be treating patients that are not newborns anymore. So the question of “is this sufficient” is indeed very relevant, and the answer – at least in mice – is “yes”.
So in 2020 we have proof by two companies that we can rescue CDD in mice, with improvements even in older mice, and the discussion has now moved to how to scale this up to work in larger brains. We have made it past stage 1.
These programs open the path for follow up therapies based on gene editing, the technology that earned the Nobel Prize for Chemistry in 2020. At the Forum we reviewed several of these programs, including a modified CRISPR approach to try to “unlock” the inactive CDKL5 in the second X chromosome of each cell, as well as “prime editing” which is a field pioneered by David Liu who explained us that CDD is one of the first diseases that they are trying to correct with prime editing. There are so many therapeutic programs in development for CDD that it would be hard to list them all in this type of update. What is important to know is that pretty much every therapeutic technology that we know already has an active program (or more) to apply it to CDD, so the question is no longer whether we will ever get a curative treatment, but instead it is “which of them” and “when”.
5. 2020 THE YEAR OF CLINICAL TRIALS (ALSO IN PLURAL) SUCCESS
Day 3 of the 2020 CDKL5 Forum came with many great news. Dr Orrin Devinsky summarized the data with Epidiolex in CDD as part of an expanded access program that showed efficacy, and then announced for the first time the results of two small investigator-initiated phase 2 studies (those are “one hospital” clinical trials) that he has led:
1. Ataluren did not have efficacy in CDD. Orrin showed us the data of a study with 18 patients, some of them with CDD some of them with Dravet syndrome, in which some patients started with ataluren and then continued with placebo and others did it the other way around (a placebo-controlled crossover study). Ataluren is a molecule that helps the cell skip premature stop codons caused by non-sense mutations, so all of these patients had non-sense mutations. Ataluren did not show an effect in reducing seizure frequency or improving cognitive, motor, or behavioral function or quality of life in subjects with either Dravet Syndrome or CDKL5 deficiency due to nonsense mutations. This randomized, placebo-controlled trial was limited by a small sample size and a treatment-phase of 12 weeks, which may be too short to identify a disease-modifying effect. .
2. But fenfluramine does have efficacy in CDD! Fenfluramine is an anti-epileptic drug that works through the serotonin system and that has been approved in the US and Europe for the treatment of Dravet syndrome, another complex neurological condition. Orrin is running a 10-patient study with fenfluramine in CDD at NYU, and was able to present the results from the first 6 patients. And the results are very good, with a 90% reduction in generalized tonic-clonic seizures that are the strongest seizure type, and also efficacy in other seizure types. He still has 4 slots open for patients with CDD ages 2 to 18, and encouraged the company developing fenfluramine to consider CDD for a phase 3 trial.
And the phase 2 study with fenfluramine was only the first out of THREE positive clinical trials in CDD that were presented that afternoon. Dr Julia Tsai from Ovid Therapeutics presented positive data with soticlestat in CDD, where a large majority of the parents reported positive improvements in different disease symptoms and where seizure improvement (the primary endpoint) continued increasing over time. Soticlestat acts through a completely novel mechanism of action that includes indirect glutamate modulation and other potential therapeutic benefits, so these different therapies are good candidates for being used in combination in the future if they get approved. Ovid also run other phase 2 studies with this drug in other genetic syndromes with epilepsy, and has not yet announced how many will be progressing to phase 3 trials or when.
And taking about phase 3 trials, the next presentation was by Alex Aimetti from Marinus who walked us through the positive results of the phase 3 trial with ganaxolone, the very first pivotal trial ever run in CDD, and that recently announced it had met its primary endpoint. The Marinus team explained how until recently the regulators were not even sure if CDD was a separate disorder, and it was thought that there were so few patients in the world that a phase 3 trial could only have 50 patients, but a couple of years later they have the Orphan Drug Designation by both the EMA and the FDA, the Rare Pediatric Designation by FDA, and a 100-patient positive Phase 3 trial in CDD. Ganaxolone led to a significant reduction in major motor seizure frequency and was well tolerated, so the company will apply for marketing authorization and plans to initiate an Expanded Access Program first in the US, that might potentially will get to additional territories later on. Marinus was awarded the 2020 CDKL5 Forum Award for the Company making a difference in the clinical space, and the company has played a unique role at helping de-risk clinical development in CDD and bringing visibility to the disorder.

I want to stress again what we just saw at the Forum: three positive clinical trials in CDD. One pivotal trial, the first ever, which was positive and will hopefully lead to the first drug approved for treating CDD, and two phase 2 studies each of which looks good enough to lead to a new phase 3 study for CDD. I don’t believe there are many rare diseases that get that type of pipeline progression in one year.
6. PREPARING FOR OBSERVATIONAL STUDIES
In the last years we have been talking much about getting the field ready for more complex studies, and by that I mean studies that will look at symptoms beyond seizures. This is particularly critical for the gene therapies. A small molecule can get away with measuring seizures and getting approved for “the treatment of seizures in CDD” even if it has efficacy beyond that symptom, but when treatments are more invasive they need to demonstrate that they have broader benefit.
In preparation for that, we had launched a series of programs including obtaining a new ICD-10-CM diagnostic code for CDD and co-hosting a PFDD meeting with FDA together with IFCR, to help identify the main symptoms that impact patients with CDD. The next step is to translate those symptoms into “clinical outcome measures” which are clinical scales that assess either one main symptom or a collection of symptoms, and that are used in trials to quantify the efficacy of treatments.
IFCR recently announced the award of an NIH U01 grant to a network of CDKL5 Clinical Centers of Excellence for the characterization over the next 5 years of a collection of clinical outcome measures that they are developing specifically for CDD by running observational studies through their network.
And the Loulou Foundation announced at the CDKL5 Forum the immediate launch of a large international observational study, in the US and Europe, in collaboration with several bio-pharma companies, to determine the feasibility of using in CDD trials a collection of clinical outcome measures that haves already been used successfully in advanced clinical trials for related disorders. The industry partners hope to be able to apply the learnings from this study to their clinical trials in the near future.
This means that the CDD community, both clinicians and patients, will soon start hearing more about the concept of observational studies and the central role that they play in getting the field ready for more complex studies.
7. BIGGER AND GLOBAL
Before concluding, I would like to highlight a couple of additional observations beyond these scientific presentations.
One is how much the idea of how rare CDD is has changed. Not so long ago it was through that we only had a couple of hundreds of patients in the world, and now we know it is one of the most common genetic causes of epilepsy. In their latest corporate presentation, Marinus now estimates about 12,500 children in the US and EU, and the actual numbers could be higher with an estimated incidence of 1 in 42,000 births.
This change in how we see CDD became particularly clear when during the Forum, the speaker from Ultragenyx, a company that was started to treat ultra-rare diseases, spoke about “serving this large patient population” in reference to CDD. And the International CDKL5 Alliance already includes 20 members, also showing the growth of the patient community and their willingness to work closer together.
And the second one is the wonderful keynote speakers that we had in the Forum. On day 1, Antonino Caridi, Ariadna’s grandfather and one of the pillars of the CDD patient community, told us about how life some time takes a turn and places you at war against a monster, and how he found in the CDKL5 Alliance a real alliance beyond just words. He spoke about how we all play a role in the movie about CDD, including the families, and encouraged all “actors” to make the best in the roles they are playing.
On day 2, Yann Le Cam, Founder of EURORDIS, told us about his own journey as a father of a now adult child with a rare disease, and the need to think global and be strategic. In his words: “think about the ecosystem, and not just your organization”. Antonino and Yann both echoed the need to respect the challenge (the monster) ahead, while creating the focus and determination to bring forward a change which requires thinking and acting globally.
And on day 3, Jeremy Levin, CEO of Ovid Therapeutics and Chairman of the Board at the Biotechnology Innovation Organization (BIO), provided us with his unique view, from his global pharma position, of what is happening in biomedical research in 2020 and how the CDD field is doing. He acknowledged the strangeness and difficulties of 2020 for everyone due to COVID-19, and the enormous challenge and disruption that it has brought to the pharma industry. Although we have seen an immediate slowdown of clinical trials, he envisions that many of the changes in trial design and regulatory policies that have been put in place to speed up the development of COVID-19 therapeutic and vaccines will also speed rare disease therapies from now on, including adopting telemedicine. These great changes are not going away, he said, they are here for the future. Jeremy also praised the CDD community for helping Marinus and Ovid deliver late-stage trial results in the middle of the pandemic.
To close, I like to end the Forum updates by reviewing the goals that the Loulou Foundation set when it started, which are to get treatments (to reach trials) by 2020 and cures by 2025. Now we know that by 2020 we’ve had not one trial, but several trials leading to three positive clinical trials including a 100-patient pivotal trial. And based on what we are seeing in the preclinical space, I believe that by 2025, we will have multiple cures (gene therapies and similar) in advanced clinical trials. As Orrin said referring to the progresses during the last year, “what we have seen these 3 days is breathtaking”. We just need five more years like this one.
I hope you enjoyed this summary! let me know your thoughts in the comments. Here are my articles on the 2018 and 2019 Forum meetings.
Ana Mingorance, PhD
Disclaimer: These are my own impressions from the presentations that I was most interested in as a scientist and patient advocate, and not an official text about the Forum by the Loulou Foundation. I write these texts with the parents of individuals with CDD in mind, so excuse also my lack of technical accuracy in parts ;-)

REPASO DEL FORO CDKL5 2020
La sexta edición del Foro CDKL5 tuvo lugar online, los días 12-14 de octubre. El Foro es una reunión anual exclusivamente por invitación que organiza la Fundación Loulou y en la que científicos y miembros de la industria farmacéutica se reúnen con representantes de la comunidad de pacientes para repasar los últimos avances en el campo. Este ha sido mi cuarto Foro CDKL5, y el tercero desde que me uní a la Fundación Loulou.
Este es un repaso para los grupos de pacientes de las principales novedades del Foro CDKL5 2020. [SPANISH VERSION - ALSO AVAILABLE IN ENGLISH]
2020 es el año en el que el mundo descubrió la urgencia por desarrollar una cura que es habitual en las enfermedades raras.

Tambien es el año en el que no hemos podido celebrar conferencias presenciales, con lo que la sexta edición del CDKL5 Foro, que hubiera sido en Londres este año, se celebró de manera virtual los días 12 a 14 de octubre de 2020. El Foro es una reunión anual que organiza la Fundación Loulou y en la que científicos y miembros de la industria farmacéutica se reúnen con representantes de la comunidad de pacientes para repasar los últimos avances el díndrome de deficiencia en CDKL5 (CDD por sus siglas en inglés). Este ha sido mi cuarto Foro CDKL5, y el tercero desde que me uní a la Fundación Loulou.
EL Doctor Orrin Devinsky, coordinador del Comité Asesor de la Fundación Loulou, resumió el Foro 2020 diciendo que “lo que hemos visto en estos tres días es extraordinario”. Para los que no estuvisteis en el congreso, os dejo con un resumen de mis conclusiones del 2020 CDKL5 Foro y lo mucho que ha avanzado la investigación en CDKL5.
Every year, the CDKL5 Foro allows us to look back in time to how the field was just a couple of years ago, review all the progresses made in the last year, and anticipate how the next year of research into CDD and drug development is likely to look like. And if we are to learn from our predictions from the past years, we will probably underestimate how much everything will move in just 12 months.
1. BIOLOGÍA INCREIBLE
El Foro siempre comienza con un repaso de lo que sabemos sobre la proteína CDKL5 y del por qué la falta de CDKL5 lleva a una enfermedad tan severa.
Yo soy bióloga celular, y estoy acostumbrada a pensar en enfermedades en función de lo que hace la proteía que falta o que tienen mutada. Por ejemplo “se trata de un canal iónico, así que cuando falta el canal las neuronas están hiper-excitables”, o “es una proteína esencial para la degradación de otras proteínas con lo que cuando está mutada se acumulan proteínas tóxicas que no se pueden degradar”. Pero el caso de CDKL5 me desmonta todos esos esquemas simplísticos. La proteía CDKL5 es una quinasa que está involucrada en tantos procesos celulares que el intentar entender lo que hace no solo nos está enseñando como funciona el síndrome de deficiencia en CDKL5, ¡non está enseñando biología que es nueva para la ciencia!
En palabras del Doctor Victor Faundez de la Universidad de Emory, que capturó muy bien este mensaje:
“Una mutación que nos abrió las puertas de una biología increíble.
”
Y vimos mucho de esto durante la jornada previa, cuando se reúnen los grupos de investigación financiados por la Fundación Loulou, y dirante el primer día del Foro. Científicos de todo el mundo intercambiaron resultados de cómo CDKL5 modula el esqueleto celular controlando los microtúbulos (los huesos de la célula), y por qué esto es crítico para la plasticidad neuronal y la formación de nuevas sinapsis (la conexiones entre neuronas). CDKL5 parece también jugar un papel en el núcleo celular, donde está el ADN, y nos mostraron como también regula la movilidad de los cílios, como los de las células epiteliales que rodean los ventrículos cerebrales y que hacen mover el líquido cefalorraquídeo. Y es que parece que CDKL5 es en realidad una quinasa que posiblemente controla muchas otras quinasas, con lo que se crea una red que se abre en muchas direcciones. A través de algunas de esas vías de señalización empezamos a identificar posibles puntos de entrada con fármacos que nos puedan ayudar a reducir algunas de las consecuencias de que falte CDKL5.
Dicho esto, está claro que como CDKL5 controla tantos procesos celulares diferentes va a ser muy difícil conseguir una recuperación completa de los síntomas atajando solo una de estas vías alteradas. Vamos a tener que reemplazar la proteína (o el gen CDKL5) para poder conseguir la recuperación completa. Pero las buenas noticias son que ya tenemos varios de estos tratamientos en desarrollo.
Algunas de las consecuencias de la falta de CDKL5 me recuerdan a otras enfermedades neurológicas. Por ejemplo la enfermedad neurológica causada por mutaciones en KIF1A, que también afecta a los microtúbulos. Y una publicación reciente describe la función de CDKL5 en riñones, donde marca a otras proteínas para ser ubiquitinadas (un sistema de degradación) me recuerda al síndrome de Angelman. Y el caso es que cualquiera de estas vías de señalización, de forma individual, es suficiente para producir un trastorno neurológico, pero en CDD se encuentran todas alteradas a la vez.
Y todo por esa mutación que nos abrió las puertas de una biología increíble.
2. COMPLETANDO LA CAJA DE HERRAMIENTAS
Una de las razones por las que hay tantos científicos interesados en CDKL5 es por esa biología que nos enseña. Otra razón las la disponibilidad de herramientas.

Los científicos no pueden investigar una proteína o una enfermedad si no tienen la posibilidad de aislar esa proteína, o de ver su actividad, o de modelizar la enfermedad en células o animales. Por eso una parte importante del esfuerzo de la Fundación Loulou y otras organizaciones se centra en generar este tipo de herramienta de investigación para completar esa caja de herramientas que hace falta para llevar fármacos desde células hasta ensayos clínicos. El año pasado en el Foro de 2019 hablamos mucho de identificar las herramientas que faltan y de coordinar esfuerzos para generarlas. Un año después, esos esfuerzos se notan.
Voy a destacar solamente tres de estas herramientas, aunque hubo más que fueron presentadas en el Foro:
Dan Lavery, Director Científico de la Fundación Loulou y anfitrión del Foro, anunció que el repositorio de herramientas de investigación Coriell va a empezar a distribuir en breve células iPSC de pacientes con CDD. Este es el resultado de varios años de la colaboración con el Hospital Infantil de Boston y que ha llevado a la obtención de varias líneas de iPSCs y controles isogénicos. Para los que no sois científicos: las células iPSC son células madre inducibles pluripotenciales, un tipo de célula madre que se obtiene a partir de células de la piel o sangre de una persona (en este caso de niños con CDD) y que los científicos “revierten” al estado de célula madre. Y son muy útiles para la investigación porque los científicos también pueden hacerlas avanzar en desarrollo y producir neuronas en cultivo, que sería neuronas con CDD. Y al estar disponibles a través de este repositorio será tan fácil traerlas a cualquier laboratorio para investigar CDD como hacer un pedido en Amazon.
Otra herramienta de investigación importante para entender CDKL5 es… pues CDKL5. La empresa Amicus Therapeutics ofreció compartir la proteína CDKL5 purificada con la comunidad científica, lo que abre muchas posibilidades de experimentos que hasta ahora no eran posibles porque producir y purificar CDKL5 resulta ser muy complicado. Pero Amicus tiene mucha experiencia trabajando con proteínas, y una vez más ha mostrado su interés en apoyar a la investigación en CDD más allá de a través de sus propios programas terapéuticos en desarrollo.
Y si hay algo importante pero que tiende a fallar en ciencia es la reproducibilidad de experimentos farmacológicos en animales (en ratones). Hace un año la Fundación Loulou firmó una colaboración con el Baylor Medical College y el Hospital Infantil de Texas para crear una plataforma de diferentes modelos transgénicos de CDD en los que poder evaluar (y confirmar) la eficacia de terapias de una forma objetiva y rigurosa. El trabajo está liderado por el Doctor Rodney Samaco, que recibió el premio de Laboratorio del Año en el Foro 2020 por el valor que esa batería de modelos transgénicos y tests comportamentales tendrá para el desarrollo de terapias en CDD. Y este valor ya quedó evidente en este Foro al demostrar a través de estos ratones la eficacia de la terapia génica de Ultragenix, como elaboro más abajo.
Completar la caja de herramientas de CDKL5 lleva muchos años, muchos científicos, mucha financiación y mucha colaboración. La caja de herramientas de CDKL5 es reflejo de lo mucho que ha madurado la investigación en torno a CDD.
3. CDD NO ES UNA ENFERMEDAD DE DESARROLLO NEUROLÓGICO
Pues si. Esta frase, tan provocativa como resulta, es una de las principales conclusiones de este año.
Llegamos a este Foro sabiendo que no se han encontrado defectos importantes de conexiones neuronales en CDD, o de anatomía cerebral, y tampoco hemos visto indicaciones de que pueda tratarse de una enfermedad neurodegenerative. Así que todo apuntaba a que CDKL5 se necesite más bien para el proceso constante y dinámico de formar y rehacer sinapsis. Y esto implicaría que el poder devolverle la expresión de CDKL5 al cerbero tendría beneficios aunque se trate de un cerebro maduro, mientras que en el caso de enfermedades en las que la migración neuronal está alterada, o hay muerte neuronal, solo tendríamos una ventana temporal limitada durante la que debemos devolver la proteína a las neuronas para poder ver mejoras. Pero solo teníamos esas pistas, y ninguna evidencia directa.
En el Foro aprendimos, a través de estudios en ratones, que la proteína CDKL5 está presente a niveles altos durante toda la vida, no solamente durante el desarrollo. Aprendimos también que si la quitamos de repente en el cerebro de ratones adultos, como empezándole la deficiencia en CDKL5 a ratones adultos utilizando un interruptor genético, esto resulta en la aparición de todos los mismos síntomas en estos ratones que si hubieran nacido sin la proteína. Joe Zhou de la Universidad de Pensilvania nos enseñó como esos ratones a los que les quita CDKL5 cuando ya son adultos desarrollan hiperactividad, problemas de sociabilidad y otros comportamientos del espectro autista, problemas cognitivos y mala coordinación motora – y esta es exactamente la presentación de CDD en ratones, donde es raro que aparezcan crisis epilépticas. Como el hecho de quitar CDKL5 en el cerebro adulto es suficiente para producir la enfermedad, Joe concluyó que “CDKL5 es indispensable durante toda la vida”.
¿Y que pasa en el caso opuesto? ¿Cuál es la consecuencia de devolverle CDKL5 a un cerebro que se desarrolló sin la proteína? Joe explicó que ha creado unos ratones con el interruptor opuesto, y que va a ver que pasa cuando les deja nacer sin expresar CDKL5 y se lo enciende de repente cuando son adultos. Esperamos ansiosos estos resultados. Entre tanto la primera respuesta parcial a estas preguntas nos llega de la mano de la terapia génica. Sharyl Fyffe-Maricich de Ultragenyx nos mostró cómo la administración de virus AAV que llevan el gen CDKL5 a ratones de 3 a 5 semanas de edad, que sería una edad humana de 4 a 11 años, mejora en ratones con CDD varios de los síntomas incluidos cognitivos y motores. Aunque nos aclaró que solo habían conseguido restaurar un tercio de los niveles de expresión de CDKL5, y que por tanto el rescate genético es parcial, estos experimentos nos indican que al menos en ratones el rescate incluso parcial de CDKL5 es capaz de reducir la enfermedad aún pasados los primeros años de desarrollo, y esto es la primera vez que lo podemos ver directamente.
Lo que todo esto nos dice es que CDD no es una enfermedad de neurodesarrollo, al menos no estrictamente hablando, porque no se trata de un gen / una proteína que es necesario durante el desarrollo del cerebro y luego ya no. En vez de eso, CDD sería una enfermedad del neuro-mantenimiento, en la que CDKL5 hace falta de manera constante durante toda la vida para asegurar el correcto funcionamiento neuronal. Por tanto no parece que nos enfrentemos a una ventana temporal transitoria durante el desarrollo durante la cual se necesita CDKL5 y si llegamos tarde el daño ya está hecho. Y eso nos supone una perspectiva mucho más favorable para el desarrollo de terapias, porque todos estos experimentos nos indican que CDD es una enfermedad candidata a terapia génica o de reemplazo enzimático incluso en pacientes adultos.
“CDD no es una enfermedad de neurodesarrollo, sino una enfermedad del neuro-mantenimiento, y eso nos supone una perspectiva mucho más favorable para el desarrollo de terapias.”
4. SE ACERCAN LAS TERAPIAS GENÉTICAS (EN PLURAL)
El tema de las terapias genéticas es el que dominó el segundo día del Foro. Jim Wilson, veterano del campo de terapias génicas, nos explicó como este tipo de terapia debe progresar a través de tres etapas antes de poder ser evaluada en grupos grandes de pacientes en ensayos clínicos. El les dio otros nombres, pero para hacerlas más endendibles, en mis propias palabras, las tres etapas son:
- Etapa 1 – Demostrar que podemos corregir CDD en ratones. Los primeros estudios se hacen generalmente en ratones que llevan el mismo problema genético que los pacientes, para demostrar que es posible corregir el defecto genético y tener una primera idea de si hay problemas de toxicidad. Para hacer una terapia génica, los científicos le quitan a un virus todo su ADN y lo rellenan con el gen que hace falta administrar a los pacientes, de modo que cuando el virus infecta al paciente lo que hace es llevar ese gen a las células. Y el mayor desafío de esta primera etapa es hacer bien ese “virus transgénico terapéutico” lo cual lleva varios elementos, y conseguir curar ratones con ese virus.
- Etapa 2 – Demostrar que podemos tratar cerebros más grandes. En esta etapa el virus se prueba en primates no humanos, que tienen el cerebro mucho más cercano a nuestro tamaño, para mirar toxicidad y la “biodistribución” que es la dispersión del virus. Y justamente este es el problema más común, la dispersión del virus por el cerebro (o el órgano deseado), que suele ser limitante y que hace más difíciles las enfermedades que no afectan solo a un grupo limitado de células.
- Etapa 3 – Primer ensayo en humanos. Esto es cuando la terapia llega a la clínica por primera vez, y se hace un estudio pequeño en un número limitado de pacientes para ver la tolerabilidad y toxicidad, antes de arriesgar un numero mayor de pacientes con una terapia que es aún experimental.
Jim nos explicó que el cuello de botella suele ser la segunda etapa, en la que hay que ir haciendo cambios en el virus transgénico para conseguir que llegue a funcionar en cerebros grandes. Otro factor es entender la mejor ruta de administración para facilitar también llegar a más células. Por ejemplo, en el caso de CDD, ver si habría que administrarlo directamente a cerebro o podemos ir por via intravenosa. En una sesión previa los científicos de Amicus nos explicaron como para poder llegar también a más neuronas lo que estaban haciendo era que su virus lleve no un gen “CDKL5 tal cual” sino un gen quimera que produce CDKL5 secretable de modo que las neuronas infectadas no solo producirán CDKL5 ellas, sino que harán de surtidor local de CDKL5 que podrá entrar en las neuronas de alrededor. Ellos llaman a esta estrategia “corrección cruzada” de CDKL5.
Y en ese momento llegamos a uno de los momentos mas emocionantes del congreso, cuando vimos las dos pruebas de concepto en ratones de dos empresas que están desarrollando terapias génicas para CDKL5 (la prueba de concepto es la demostración de que podemos hacer terapia génica en ratones con CDD y que funciona)
El primero en presentar fue el equipo de Jim Wilson, que está generando la terapia génica de la empresa Elaaj Bio. Ralf Schmid nos hizo un repaso de muchísimos experimentos que han realizado para optimizar el “virus transgénico terapéutico”, desde ver cual de las múltiples formas de CDKL5 es la que hace falta (hay cuatro!), a ver que pasa si expresamos demasiado CDKL5 en el cerebro (por si hay efectos de toxicidad), a probar las diferencias de recuperación entre ratones macho y hembra, ya que en las hembras (como en personas) la mitad de las neuronas está afectada y la otra mitad no. A la hora de realizar la prueba de concepto este grupo eligió ir a por el escenario perfecto, que es tratar ratones con CDD el mismo día que nacen. Eso asegura que el virus llegue mejor a todo el cerebro y que el ratón sea tratado lo antes posible. Y la prueba de concepto del escenario perfecto salió espectacular. Ralf nos enseñó que con su terapia génica son capaces de devolver CDKL5 a los niveles normales, tanto en expresión como en actividad (sabemos ver la actividad mirando una diana de CDKL5 llamada EB2). Nos enseñó imágenes preciosas de la expresión de CDKL5 por todo el cerebro, y como son capaces de corregir todos los síntomas de los ratones tras administrarles la terapia génica directamente en el líquido cefalorraquídeo. Incluso nos enseñó mejoras en el EEG preliminares, y estos resultados los han replicado en un laboratorio independiente usando dos tipos de ratón transgénico adicionales que llevan otras mutaciones en CDKL5. Es difícil hacer ciencia mejor hecha y más robusta que la que nos enseñó este grupo.
Y ahora están optimizando este virus para esa etapa 2 de la que nos hablaba Jim que consiste en ver cómo conseguir que tenga buena biodistribución en el cerebro grande de primates para así poder evaluar su posible toxicidad y la mejor dosis a usar y poder entonces pensar en ensayos clínicos.
Y como os adelantaba antes, Ultragenyx también nos presentó su prueba de concepto en ratones con su propia terapia génica por la que recibieron en este Foro el premio a la empresa que marca una diferencia en el espacio preclínico. Fue muy interesante que Ultragenyx optó por diseñar la prueba de concepto diferente a la del grupo anterior. En vez de ir a por el escenario perfecto, fueron a por el escenario realista, en el que trataron ratones mayores sabiendo que solo llegarían a rescatar parte de la producción de CDKL5. Y Sharyl nos enseñó como incuso ese rescate parcial con el virus también administrado directamente en el líquido cefalorraquídeo pero a ratones mayores, de edad humana equivalente a 4 a 11 años, también resultaba en eficacia en diferentes síntomas. Aunque no tan espectaculares como tratando ratones recién nacidos, estos resultados son tremendamente esperanzadores porque en los ensayos clínicos muy posiblemente nos quedemos también con producción parcial de la proteína (ellos llegan a un 35% de los niveles), y la mayoría de los casos no serán en recién nacidos. Así que esa pregunta que se hace Ultragenix de “¿es el rescate parcial suficiente?” es ciertamente relevante, y la respuesta parece ser, al menos en ratones, un si.
Así que 2020 nos ha traído dos pruebas de concepto de dos empresas diferentes de que el rescate genético de CDD en ratones es posible, inclusyo en ratones “mayores”, y ahora la discusión avanza a esa etapa 2 de ver como escalar los experimentos para cerebros mayores. ¡Etapa uno superada!
Y estos proyectos de terapia génica abren la puerta a otras terapias basadas en la edición genética, que es la tecnología que ha ganado el Premio Nobel de Química este año. Y en el Foro vimos varios de estos programas, incluido uno para utilizar una estrategia de CRISPR modificado para “activar” la copia de CDKL5 que está en segundo cromosoma X y que se encuentra inactivo, así como estrategias de “prime editing” que es un campo que lidera David Liu quien nos explicó que CDD es de las primeras enfermedades para las que se está buscando aplicación terapéutica de esta nueva tecnología que es una especie de CRISPR 3.0. Hay tantos tipos de estrategia que sería difícil cubrirlas todas en un resumen de este tipo, pero lo que importa es que sepáis que prácticamente todas las modalidades de tecnología de terapia se está aplicando ya a buscar una terapia para CDD, y que la pregunta ya no es si tendremos algún día una de estas terapias curativas disponibles. La pregunta es mas bien “cuál de ellas” y “cuándo”.
5. 2020 EL AÑO DEL ÉXITO EN LOS ENSAYOS CLÍNICOS (EN PLURAL)
El día 3 del Foro nos llegó cargado de noticias. El Dr Orrin Devisnky empezó repasando los datos con Epidiolex en CDD de los ensayos en abierto que muestran eficacia, y luego nos anunció por primera vez los resultados de dos ensayos clínicos de fase 2 que han realizado en la Universidad de Nueva York (NYU):
1. Ataluren no mostró eficacia CDD. Orrin anunció los resultados de un ensayo clínico que han realizado en su hospital con 18 niños con Dravet o deficiencia en CDKL5 causado por mutaciones non-sense, ya que ataluren es un fármaco que hace a la célula saltarse las mutaciones non-sense. Ataluren no mostró eficacia en cuanto a reducción de crisis ni a mejoras de comportamiento o cognitivas en niños con Dravet o deficiencia en CDKL5 causado por mutaciones non-sense. Como el ensayo clínico fue en un numero pequeño de pacientes, y solo durante 12 semanas, es posible que la duración fuera insuficiente para poder ver eficacia con un fármaco que se espera que actúe sobre la enfermedad (no los síntomas).
2. Pero fenfluramina si que funciona en CDD! Fenfluramina es un fármaco que actúa sobre el sistema de serotonina y que está aprobado en EEUU y recientemente en Europa para el síndrome de Dravet, otra enfermedad neurológica compleja. Orrin está realizando un ensayo clínico de fase 2 con 10 pacientes con CDD; de los cuales ya han concluido 6 y nos anunció esos resultados por primera vez en el Foro. De esos 6 pacientes, la reducción en la frecuencia de crisis tónico-clónicas generalizadas (el tipo de crisis más fuerte) fue del 90%, y también vieron reducciones en otros tipos de crisis. Orrin por tanto animaba a la empresa propietaria de fenfluramina a considerar CDD para un ensayo clínico de fase 3.
Y el ensayo de fase 2 con fenfluramina fue solo el primero de TRES ensayos clínicos con resultados positivos en CDD que vimos esta tarde. La Doctora Julia Tsai de Ovid Therapeutics nos presentó los datos positivos obtenidos con Soticlestato/TAK-935, con el que la mayoría de las familias reportó mejora en muchso de los síntomas y se constató una bajada en la frecuencia de crisis (el objetivo del ensayo clínico) que fue en aumento a medida que pasaban las semanas. EL soticlestato actúa por un mecanismo de acción nuevo que incluye la modulación indirecta del sistema de glutamato entre otros, y que por tanto podría ser combinado con otros de los fármacos en desarrollo para CDD si llegan a ser aprobados. Ovid también ha realizado ensayos de fase 2 en otros tres síndromes con epilepsia y aún no han anunciado cual de ellos avanzará a ensayos clínicos de fase 3.
Y hablando de fase 3, la última presentación fue de Alex Aimetti de Marinus que nos hico un repaso de los resultado de su ensayo clínico de fase 3 con ganaxolona en CDD. Este es el primer ensayo “final” (de fase 3) que hacemos en CDD, y recientemente anunciaron los resultados positivos. El equipo de Marinus nos recordó como hace a penas unos años las agencias reguladoras del medicamento no creían que CDD fuera una enfermedad aparte, y se dudaba de que fuera posible reclutar mas de 50 pacientes en un ensayo clínico porque creían que había muy pocos en el mundo. Y en un par de años han conseguido la designación de fármaco huérfano por la FDA y la EMA, la designación de enfermedad rara pediátrica por la FDA, y completar un rensayo clínico con 100 pacientes con éxito. En este ensayo ganaxolona llevó a una reducción en la frecuencia de crisis significativa y la tolerabilidad fue favorable, con lo que la empresa pedirá la autorización para sacar el fármaco al mercado en Estados Unidos y Europa. Además van a comenzar un programa de “expanded Access” (de uso compasivo) en Estados Unidos, en vista de posteriormente ampliarlo a otros territorios. Marinus recibió en el Foro 2020 el premio de empresa que marca una diferencia en el espacio clínico, y es una empresa que ha jugado un papel crucial en visibilizar la enfermedad y mostrar la viabilidad de desarrollar terapias para CDD, abriendo las puertas para todos los ensayos que siguen.

Quiero asegurarme de que destaco lo que vimos ese día: tres ensayos clínicos con resultados positivos en CDD. Uno de fase 3, el primero para esta enfermedad y que esperamos que se convierta en el primer fármaco aprobado para CDD, y dos de fase 2 ambos con resultados suficientemente buenos como para llevar esos fármacos a ensayos de fase 3. No creo que haya muchas enfermedades raras que tengan una progresión de su “pipeline” de ensayos clínicos tan buena en un solo año.
6. PREPARÁNDONOS PARA LOS ENSAYOS OBSERVACIONALES
En los últimos años hemos hablado mucho de prepararnos para los ensayos clínicos más complejos, y por ello me refiero a los ensayos clínicos que miran más síntomas que solamente la epilepsia. Porque en un ensayo clínico con un fármaco de los clásicos es suficiente con medir eficacia en las crisis para pedir la aprobación, pero con los tratamientos más invasivos como las terapias génicas hace falta mostrar eficacia mucho más amplia para poder justificar su probación.
Para prepararnos para esos ensayos complejos lanzamos una serie de proyectos como fueron pedir un código ICD-10 para CDD el año pasado, o la celebración de un PFDD (una reunión) con la FDA americana para identificar el tipo de síntomas que tiene mayor impacto para las familias. Y el paso siguiente es traducir esos síntomas en escalas clínicas que se puedan usar en ensayos clínicos para cuantificar la eficacia de las terapias.
El grupo de pacientes americano, IFCR, recientemente anunció que su red de Centros de Excelencia ha obtenido un proyecto financiado por el gobierno americano (el NIH) para caracterizar una serie de escalas clínicas que están desarrollando específicamente para CDD, y que validarán a través de una serie de ensayos observacionales en sus hospitales.
Y la Fundación Loulou anunció en el Foro CDKL5 el comienzo en los próximos meses de un estudio observacional internacional que tendrá lugar en Estados Unidos y Europa, realizado en colaboración con un grupo de empresas farmacéuticas, para estudiar la posibilidad de usar en CDD una seria de escalas médicas que ya han sido utilizadas con éxito en ensayos clínicos en otras enfermedades similares. Las empresas colaboradoras esperan poder usar las observaciones de este estudio para diseñar sus ensayos clínicos en el futuro próximo.
Así que en los meses que viene la comunidad CDD, tanto médicos como pacientes, recibirán más información sobre el concepto de ensayos observacionales y el papel que juegan en informar sobre el diseño de ensayos clínicos más complejos.
7. MÁS Y GLOBALES
Antes de terminar este resumen quiero destacar un par de observaciones que no son avances terapéuticos.
La primera es lo mucho que ha cambiado nuestra idea de cómo de raro es CDD. Hasta hace no tanto pensábamos que había un par de cientos de pacientes en el mundo, y hoy sabemos que es una de las causas genéticas más comunes de epilepsia. En su última presentación corporativa, Marinus habla de que hay unos 12.500 niños entre Estados Unidos y Europa con la enfermedad – y la realidad es que serán más, basado en la incidencia de 1 en 42.000 nacimientos.
Y este cambio en cómo vemos la rareza de CDD quedó particularmente de manifiesto en el Foro cuando una investigadora de Ultragenyx, que es una empresa especializada en enfermedades ultra-raras, se refirió a CDD diciendo que se preparan para “servir a esta población tan grande de pacientes”. Y la Alianza Internacional de CDKL5 ya tiene 20 miembros, lo que muestra el crecimiento y la unidad de la comunidad de pacientes.
Y el segundo tema a destacar es la calidad de los ponentes destacados (keynote) que trajo el Foro este año. El primer día el ponente destacado fue Antonino Caridi, abuelo de Ariadna y uno de los pilares de la comunidad de pacientes, quien nos contó como a veces la vida tuerce el camino y nos enfrenta con un monstruo, y como el ha encontrado personalmente en la Alianza CDKL5 mucha más alianza que solo en el nombre. Nos habló de como en la película de CDKL5 cada uno tiene un papel, incluidas las familias, y nos instaba a hacer lo mejor que podamos el papel que nos ha tocado a cada uno.
El segundo día, el ponente invitado fue Yann Le Cam, fundador de EURORDIS, quien nos contó su propio camino como padre de una niña (ahora adulta) con una enfermedad rara, y nos habló de la necesidad de pensar de forma global, y de ser estratégicos. Tanto Atonino como Yann hicieron referencia a esa necesidad de respetar el desafío (el monstruo) que tenemos ante nosotros, a la vez que debemos crear el foco y determinación que necesitamos para poder crear un cambio que requiere que pensemos y actuemos a escala global.
Y el tercer día, Jeremy Levin, Director Ejecutivo de Ovid y Presidente de la Junta de la Biotechnology Innovation Organization (BIO), nos ofreció su perspectiva única, desde su visión de la industria farmacéutica a nivel global, de lo que está pasando con la investigación en biomedicina en 2020 y cómo ve el desarrollo de terapias para CDD. Empezó reconociendo lo raro y difícil que es 2020 para todos, por culpa de COVID-19, y el enorme desafío y disrupción que ha creado para la industria farmacéutica. Aunque a corto plazo hemos visto una desaceleración en los ensayos clínicos para todas las otras enfermedades, Jeremy cree que los cambios en diseños de ensayos clínicos y normativas de desarrollo de fármacos que se han adaptado para acelerar el desarrollo de tratamientos y vacunas para la COVID-19 va a ayudar también a acelerar la velocidad de desarrollo de fármacos para enfermedades raras, como por ejemplo la adopción extendida de la telemedicina. Estos cambios no se van a ir, nos dio, están aquí para quedarse. Jeremy también destacó el papel de la comunidad CDD para asegurar que los ensayos clínicos de Ovid y Marinus pudieran ser completados en medio de una pandemia.
Para acabar, me gusta terminar mis repasos del último Foro recordando los objetivos que se marcó la Fundación Loulou cuando empezó, que fueron: tener tratamientos en ensayos clínicos en 2020, y curas en ensayos clínicos en 2025. Ahora que es 2020 podemos decir que no es que hayamos tenido un ensayo clínico, es que hemos tenido varios y tres han dado positivo, incluido un ensayo de fase 3 / fase final con 100 pacientes. Y en base a lo que estamos viendo en el espacio preclínico, yo creo que para 2025 tendremos los ensayos clínicos con terapias génicas en estadíos muy avanzados. Como Orrin dijo en referencia a los progresos en el último año, “lo que hemos visto en estos tres días es extraordinario”. Solo nos hacen falta 5 años más como este.
Espero que os haya gustado el resumen. Ya me diréis lo que os parece en los comentarios. Y os dejo también el enlace al resumen del Foro de 2019 y de 2018 (en inglés).
Ana Mingorance PhD
Nota: este texto captura mis impresiones de las presentaciones del Foro que más me interesaron como científico y como defensora de los pacientes, no es un texto oficial del congreso emitido por la Fundación Loulou. Escribo estos resúmenes para los padres de personas con CDD, así que a veces me tomo ciertas licencias a la hora de explicar las partes mas técnicas ;-)

Gene therapy for Dravet syndrome – 2020 update
There are multiple gene therapy and oligonucleotide programs in development for Dravet syndrome including those that supply and extra copy of the SCN1A gene and those that boost expression from the healthy SCN1A gene copy. Clinical trials have already started, with Stoke Therapeutics initiating the first clinical trial with a disease-targeting therapy in Dravet syndrome in summer 2020. Behind Stoke, gene therapies are approaching the clinic with Encoded Therapeutics having the most advanced clinical candidate and preparing for trials in 2021.
In February of 2019 I reviewed the state of development of gene therapy approaches for Dravet syndrome at that time. A lot has changed in a year a half, for good. So I felt this update was long due.
Here is a review of the gene therapies in development for treating Dravet syndrome, how each of them works, and the timings that we anticipate for clinical trials.
CURRENT GENE THERAPIES IN DEVELOPMENT FOR DRAVET SYNDROME
In diseases like Dravet syndrome where the problem is that a copy of the gene is missing or not functional due to mutations, the desired therapy is one that can restore normal gene expression and therefore normal protein production. In other words, we need more protein.
In the case of Dravet syndrome, the gene is SCN1A, and the protein that is needed is the neuronal sodium channel Nav1.1. As a result of mutations in the gene, the number of Nav1.1 channels at the neuronal surface is not sufficient, there is less sodium crossing the membrane, and the neuron cannot fire properly. The result is Dravet syndrome.
One particularity of Dravet syndrome is that only one of the two copies of the SCN1A gene is affected, the second one is perfectly fine, so that second copy can serve as the supply for extra protein production. As you will see, the most advanced programs are exploiting this possibility.
Broadly speaking, there are two potential approaches to restore protein expression in Dravet syndrome: you either supply the cell with an extra healthy copy the gene, which will lead to more protein being produced, or you try to boost the expression from the healthy gene.
(1) Supply a new copy of SCN1A
When people think about “gene therapy”, the type of therapy they are thinking about is the one where the DNA of a virus gets replaced by the gene that the person needs, and that modified virus is used as a Trojan horse to infect cells and deliver them the therapeutic gene.
The most commonly used virus for gene therapy is the Adeno-Associated Virus (AAV), and because we have so much experience with gene therapies being developed and approved using AAV, this virus is the first choice for most in the gene therapy space. But AAVs are small virus, and have a limited capacity for the size of genes that they can carry, and SCN1A happens to be way too large for using these virus to carry the gene. In January of 2018, before we knew about the new gene therapies currently in development for Dravet syndrome, I reviewed this problem in the article “big gene, small virus”.
One way around this is to use other viruses that are larger and can therefore carry larger genes inside. One of these is the Adenovirus, which we all hear much about recently because it is also the virus of choice for the AztraZeneca vaccine in development for COVID-19.
|| The Spain-France-Israel consortium CureDravet is developing a gene therapy for Dravet syndrome using Adenovirus, a type of high-capacity virus that is large enough to contain the entire SCN1A gene. The leading Spanish group from Rubén Hernández has presented early results at conferences (e.g. European Paediatric Neurology Society (EPNS) 2019) showing rescue of multiple disease phenotypes in transgenic mice by administration of an adenovirus-based SCN1A gene therapy. As of the last public update this is still a preclinical program working towards the development of a clinical candidate.
Another way around the gene-vs-virus size issue is to use not one, but two AAVs, and to make each carry half of the SCN1A gene. This is a strategy that is also being explored for gene editing using CRISPR because the CRISPR approach requires expressing in the tissue proteins whose genes are too large to fit into an AAV. So the strategy of two AAVs is being explored both for rare genetic diseases with large genes such as Dravet syndrome, and for gene editing in general.
|| At UCL, the team of Rajvinder Karda and Simon Waddington is working on two approaches. One is to use another type of large-capacity virus, Lentivirus, to carry the SCN1A gene. The second approach uses two AAV virus, each containing half of the SCN1A gene, which are able to recreate the full channel once they co-infect the same cells. As of the last public updates these are also still preclinical programs working towards the development of clinical candidates.
(2) Boost expression of SCN1A
Another strategy to restore Nav1.1 levels is to target the good SCN1A gene copy or some stem downstream of it to increase protein production, without needing to add an external gene copy with a virus. This one is the strategy most advanced for Dravet syndrome.
There are several ways to do this, and luckily for us many of these approaches are being pursued and some of them are already in clinical development or getting very close.
As a small biology primer: genes (like SCN1A) are large stretches of DNA that contain the information needed to produce proteins (like Nav1.1). These large stretches of DNA are first copied into large stretches of RNA (pre-messenger RNA or pre-mRNA), then some sections that are not meant to be part of the final protein are removed from the RNA to make it shorter (mRNA), and then the mRNA is read to produce the final Nav1.1 protein.
What happens in Dravet syndrome as a result of having a mutation (or deletion) in one copy of the SCN1A is the following:

By now we know that the levels of Nav1.1 protein production are regulated in the cell at different levels along this sequence of steps, offering scientists multiple points where to act when trying to boost production of Nav1.1:
Regulation at the step of gene expression 1: the SCN1A gene has sequences around it that tell the cell how much the gene should be copied and therefore how much RNA should be produced. These are promoter or enhancer or other regulatory regions that could be targeted to boost gene expression and protein production.
Regulation at the step of gene expression 2: cells produce some RNAs that are not meant to produce proteins, but to match the sequence of some genes and prevent them from being read by the cell. SCN1A happens to be the target of one of those Natural Antisense Transcripts, or NAT. This endogenous “repressor” of SCN1A expression could be also targeted to boost gene expression and protein production.
Regulation at the step of RNA processing: neurons produce a large amount of pre-mRNA of SCN1A, with only a fraction of it being processed into mRNA which is the final form that will be used to produce proteins. This creates a “reserve pool” of pre-mRNA that could potentially also be targeted to get more productive mature mRNA and therefore boost protein production.

The first company to try one of these approaches was OPKO Health. OPKO decided to go after the Natural Antisense Transcripts that limits SCN1A expression by developing an oligonucleotide that could displace it, releasing the good SCN1A copy and leading to more mRNA and more Nav1.1 protein levels. Because their oligonucleotide antagonizes the NAT for SCN1A they called it an AntagoNAT (OPK88001, previously CUR-1916). While OPKO indicated plans to initiate clinical trials as early as in 2017, these timelines were moved to 2018 and then 2019 and as of summer 2020 there are no news of whether this program is still active. In the meantime, other corporate programs have appeared and progressed further into the clinic.
As a side note: oligonucleotide therapeutics are NOT gene therapies, if we stick to the actual meaning of gene therapies using genes as therapies. But because oligonucleotide therapeutics target gene expression or mRNA processing leading to more protein production we often bundle them together with the “real gene therapies”. Both are approaches that target the genetic problem in the disease, either correcting it or compensating for it (like when boosting the healthy gene copy expression). Both are expected to result in increased levels of Nav1.1. And both are expected to lead to transformational improvements across different symptom domains in patients. So for all purposes it makes sense to talk about together when reviewing gene therapies.
2018 brought the good news that Stoke Therapeutics was developing an antisense oligonucleotide treatment to boost expression of SCN1A as well. This oligonucleotide binds to the pre-mRNA, facilitating the transformation of some of the pre-mRNA “reserve pool” into mature mRNA and leading to more Nav1.1 protein. Stoke recently published their preclinical proof of concept (July 2020 and August 2020), showing how they were able to produce an antisense oligonucleotide able to trigger this pre-mRNA to mRNA processing specifically for SCN1A and not affecting other sequence-related sodium channel genes. Their therapeutic candidate, called STK-001, increased expression of the SCN1A mRNA and Nav1.1 protein in mice, and had remarkable efficacy in mice with Dravet syndrome caused by SCN1A haploinsufficiency.
|| Since my early 2019 update, Stoke Therapeutics had an IPO, obtained an FDA Orphan Drug Designation for STK-001, and in August of 2020 it initiated a Phase 1/2s study in patients 2 to 18 years with Dravet syndrome. The dosing of the first patient in this study represented a historical moment for the Dravet syndrome field: for the first time, a person born with Dravet syndrome was given a therapy designed to correct their genetic problem (SCN1A haploinsufficiency). That day the field moved from developing and testing symptomatic treatments to developing and testing disease-targeting treatments, which the patient community often prefers to call cures.
The program from Stoke Therapeutics is the most advanced of all gene therapies and gene therapy-like approaches in development for Dravet syndrome. The second program in line is a gene therapy approach by Encoded Therapeutics that was still in stealth mode in 2018.
|| In June 2019, Encoded Therapeutics emerged from stealth mode with a series C of $104M with a lead program for Dravet syndrome. Encoded managed to beat the gene-vs-virus size problem by using AAV, the gold-standard viral vector for gene therapy, to deliver to the brain a synthetic transcription factor for SCN1A. In other words, the SCN1A gene might be too large, but we can still AAV to deliver to the brain the gene of a specific SCN1A booster. In barely one year, Encoded has closed another funding round of $135M, obtained the Orphan Drug Designation and Rare Pediatric Disease Designation by the FDA for their ETX101 gene therapy for Dravet syndrome, and anticipates to start clinical trials in 2021.
Several academic efforts follow Stoke and Encoded in the pursue of boosting expression from the good SCN1A gene copy. All of these projects are in early preclinical stages, and they have not yet published a solid proof of concept in a Dravet syndrome mouse model, which is an initial stage prior to advancing the treatments towards clinical trials. These programs are therefore all years away from clinical trial initiation, with no guarantee of succeeding.
|| An academic group in Italy, with funding from CURE and the Dravet Syndrome European Federation, is researching an oligonucleotide approach to boost production of Nav1.1 protein though a combination of transcriptional and translational activity. There have been no recent public updates on the progress of this project.
|| The Vania Broccoli lab in Italy in collaboration with Gabriele Lignani lab at UCL are using a CRISPR approach to target the promoter of SCN1A and increase its activity, therefore boosting production of Nav1.1 from the good SCN1A gene copy. They use a variant of CRISPR that uses dead Cas9 to find – but not to cut – the desired genomic region and activate it. They have demonstrated the ability of upregulating Nav1.1 using this system using lentivirus in vitro, and published an early proof of principle in mice by using two AAVs to deliver all of the different elements of the CRISPR system into the brain, which would not otherwise fit into one AAV. It is early to know if these dual-AAV approaches will achieve sufficient biodistribution and expression in the brain, or if single-AAV or single-Adenovirus approaches are needed, probably reducing the size of the cargo like Encoded is doing with their transcription factor approach.
|| The Yamakawa lab in Japan has also published a recent study using CRISPR with dead Cas9 to increase SCN1A gene expression and protein production in mice. For multiple reasons, their approach cannot be considered a proof of concept for the future clinical treatment. For example they used a type of AAV (PHP.B), which has fantastic biodistribution in mice but not in primates, and administered it via intravenous administration. Peripheral administration of AAV to target neurological diseases also seems an approach less viable in patients than in mice. In this case part of the CRISPR system was genetically encoded in the mice, so they only needed administration of one AAV. This would also not be possible in patients, who would need the entire treatment to be delivered to them using suitable route of administration and virus type. But what this study provides is a proof of biologic rescue of SCN1A expression in GABAergic neurons starting at 4 weeks of age in mice, and the improvement in seizures, mortality and behavioral phenotypes that result from such rescue.
|| SCN1A encodes for the alpha subunit of Nav1.1, so less alpha subunit means less mature sodium channels, but there is also a beta subunit involved in channel formation which is encoded by SCN1B. The Hampson lab in Toronto recently published a gene therapy approach using AAV to deliver an additional copy of SCN1B, which is a small gene that fits the virus. The reasoning is that upregulating the beta subunit might be able to drag more alpha subunits to the surface and result in more total Nav1.1. In the study, funded by Dravet Canada and the US Dravet Syndrome Foundation, mice heterozygous for SCN1A had improvements in mortality, seizures and behavioral outcomes, although some of these were more pronounced in males or females. None of the SCN1A boosting approaches had reported any sex-bias when it comes to efficacy. The beta subunit produced by SCN1B is auxiliary to multiple channels, not unique to Nav1.1, and the study did not determine which of these channels might be mediating efficacy, so the relevance of this study to the SCN1A-targeted therapeutics is unclear.
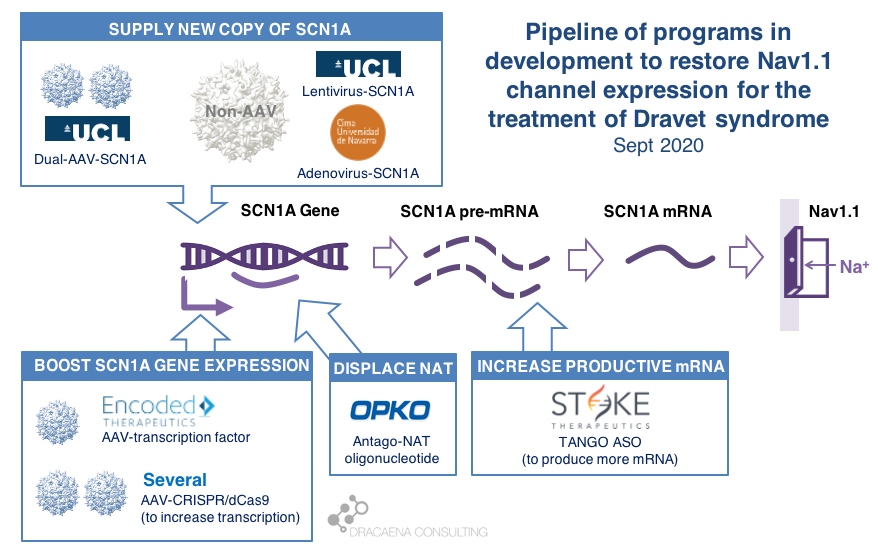
FINAL THOUGHTS
There are several differences between oligonucleotide treatments and gene therapies. Some of these are differences that are more important for us scientists. For example to better understand the need to increase SCN1A expression in all cells that naturally express the gene (what oligonucleotides do) versus the convenience of boosting it only in those that are the most affected in the disease (an option available only for gene therapies because they can use cell-specific promoters). Other differences are more important to patient families, such as the need to repeat dosing with oligonucleotide treatments, usually spaced out by several months, versus the once-and-done single-administration approach of the gene therapies. In the end what we all want to see is which approach produces the most clinical improvement with the least side effects and burden of administration, and it is very likely that we will want to have multiple options in the market.
When thinking about these options, the field of SMA comes to mind. Spinraza (by Biogen) is an ASO able to increase the production of SMN, and a total game-changer in a terrible disease that used to kill babies. Then Zolgensma (by Novartis) reached the market, as the first gene therapy using AAV to deliver a copy of the SMN1 gene. With both in the market we have started seeing clinical studies to evaluate the combination of the ASO and the gene therapy in patients with SMA. And last month FDA approved risdiplam (Evrysdi) from Genentech/Roche which is a small molecule that can be taken orally and that increases the production of SMN. In 4 years the field of SMA has gone from facing a death sentence to having three very different modalities all able to rescue the missing protein expression (ensuring access to these treatments is a different topic, but I still want to highlight that there is a lot of work to do there).
So while we keep an eye on Stoke and Encoded and the development of their two very different approaches, we might want to keep another eye following the developing of small molecule activators of the Nav1.1 channel, for which there are several programs in the development.
We also want to make sure that the current oligonucleotide treatments and gene therapies in development are successful. A major challenge they face is that we don’t know how to design a clinical trial with a gene therapy in Dravet syndrome. So far clinical trials measuring seizure frequency have been very successful, but a gene therapy is expected to improve the syndrome beyond just seizure frequency. The field of Dravet syndrome is still immature when it comes to clinical outcome measure development and validation for non-seizure outcomes (for non-scientists in the audience: we don’t know how to quantify improvements of the disease in a clinical trial beyond seizures). Both Stoke and Encoded are running observational studies that will hopefully identify the best outcome measures and endpoints that will be needed for pivotal studies.
And another development that I miss is that despite so many approaches trying to increase the levels of Nav1.1 we don’t have any biomarker that could help us measure the levels of functional or total Nav1.1 in patients. This will make it hard to interpret clinical trials where a dose of the treatment is ineffective based on the selected outcome measures. And having a biomarker for protein levels will provide early data to encourage longer patient monitoring as we wait for some of those outcome measures to show enough of a change. So in parallel to a race for developing new treatments, we are seeing a race to de-risk the field fast enough so that those treatments get their best shot at succeeding in a pivotal trial.
IN SUMMARY
There are multiple gene therapy and oligonucleotide programs in development for Dravet syndrome including those that supply and extra copy of the SCN1A gene and those that boost expression from the healthy SCN1A gene copy.
Clinical trials have already started, with Stoke Therapeutics initiating the first clinical trial with a disease-targeting therapy in Dravet syndrome in summer 2020.
Behind Stoke, gene therapies are approaching the clinic with Encoded Therapeutics having the most advanced clinical candidate and preparing for trials in 2021.
I look forward to seeing precompetitive collaborations around the common challenges of validating clinical outcome measures and biomarkers, which are needed to maximize the success of gene therapies for Dravet syndrome.
Ana Mingorance PhD

Your rare disease doesn’t have an ICD-10 code. Now what?
The rare genetic disease CDKL5 Deficiency Disorder (CDD) has been designated with a new disease code in the International Classification of Diseases (ICD), the medical classification list from the World Health Organization (WHO). The CDKL5 Deficiency Disorder diagnostic code will be incorporated in the October 1, 2020 classification revision.
This article covers what is an ICD-10, why it is important, and how patient communities can apply to have a code created for their disease.
Today we have a press release explaining that CDKL5 Deficiency Disorder has been granted an ICD-10 diagnostic code, and that it will become effective October 1 2020.
This was quite a major achievement for us. Let me explain you why it matters and how we did it.
WHAT ARE THE ICD-10 CODES
Imagine I give you a large box, and you cannot see what’s inside. I only give you a list of descriptive cues:
- Furniture
- Wood
- Four legs
- No seatback
With the last cue we can now rule out a chair, but it could still a table or a seat stool. This is not the Taboo game where your partner has to guess a word and you have to describe it using other words. This is rather a game of “you can only use words from the dictionary” and a dictionary that has no word for table.
Believe it or not this is the daily life of clinicians when they see patients with rare diseases. While rare diseases have names, these are often not included in the International Classification of Diseases, and without a code for them, clinicians need to use other codes to describe their patients, and it ends up looking as if they are just giving cues.
The International Classification of Diseases is the medical classification list from the WHO, that uses alphanumeric codes to ensure we can call the same disease the same way, no matter the country or the language.
But a lot of the rare diseases are missing in that classification.
Until now, the international codes (ICD-10 codes) for a child with CDKL5 deficiency disorder might look like this:
- F88 Global developmental delay
- G40.411 Epilepsy, refractory, with generalized seizures
- H47.61 Cortical visual impairment
- R28.2 Apraxia (when you have a motor impairment due to a brain – not muscular- problem)
- Q99.8 Genetic abnormality
- and so on
And this is happening exactly like this for a lot of rare diseases: clinicians have to almost use cues, using the symptoms and diagnoses for which there are codes, because the name of the disease is not in the dictionary and they have to stick to words in the dictionary.
Now imagine that there are over one thousand cases with CDKL5 deficiency (CDD) around the word, all coded with some variations of descriptive codes as above. And now I sit you in front of the medical database and ask you the following questions:
How many patients are out there with CDD? You don’t know, because you cannot perform a search since there is no common code
How many children are born per year and what is their life expectancy? Same, no idea because we cannot track them
Should this particular child have access to occupational therapy? You might not know if the physician didn’t use the right codes that give access to it, but if we had a code for CDD the answer could only be “yes, absolutely”
And should this child have access to ganaxolone once approved? Not sure, because while regulatory agencies recognize CDD as a stand alone disease, the international medical classification does not.
As you can see this is one problem with a lot of consequences.
I am telling you all this because we have changed it. Starting October 1 2020, when the next revision of the ICD-10 dictionary becomes effective, there will be a unique code to designate CDD and that is G40.42.
From October we won’t need to use a list of cues to tiptoe around the name-less disorder, we will be able to code into the medical record the disease for what it is. We will know how many people are out there because it will be possible to search how many people have that code in their medical records, and where they are, and make sure they have access to the services and therapies that they are entitled to.
This was a big big milestone, and if your rare disease has no ICD-10 code and you want to understand how we did it, keep reading below.
THE APPLICATION PROCESS
The country where these ICD-10 codes are most important is the US because of the insurance and reimbursement system, which requires the use of specific codes. There it is called the ICD-10-CM, for Clinical Modification. The revision of the dictionary is done once a year, every October 1, and it is managed by a division of the National Center for Health Statistics (under the CDC).
There are two reviews a year, and in order to get into the agenda you have to submit an application 3 months before. For example the next review for new code proposals is in March 2020, and the deadline was December 6 2019.
We applied in June 2019, and got accepted to be included in the review that took place in September 10-11. This was followed by an open consultation period of two more months, during which you are encouraged to secure support letters, and then we got the news 2+ months after that. Since we started preparing our application around March (2 months before the application deadline), the entire process took 10 months and remember that the effective date is October of the following year, so this is not something you can do in a rush.
STEP 1 – PROPOSAL
The proposal itself is not hard. It was about 2 pages. I wrote it following very useful advice from others who had gone through the process before (see section below), and it was submitted as a joint application from us (Loulou Foundation) and the IFCR, with the support of the US CDKL5 Centers of Excellence. This helped convey the message that having a unique ICD-10 for the disease was of critical importance for the clinicians treating these patients.
The 2 pages essentially argued that CDD is a unique disorder (not under another disease code), with unique medical needs (not identical to other diseases), and that not having a code was introducing variability in coding between patients that was hurting patient care and access to therapies.
The difficult part is what comes after those arguments, and that is what code you are proposing and why. Because you just don’t say “hey we need you to create a code” but a proper “we need a code for the disease, and this is how the new code should look because it belongs to this X section of the coding dictionary”.
That was our headache for those months. We are talking about a developmental and epileptic encephalopathy with a monogenetic cause. So it fits in three places in the classification:
- It could be under neurological conditions
- It could be under conditions with epilepsy
- It could be under genetic conditions
At the research Foundations we are not experts in medical coding, and clinical practitioners are not either, they are users of the medical coding. So we had to talk to people at the leading medical organizations (like the American Epilepsy Society and the American Academy of Neurology) who specialize in medical coding to help us see which code to propose. The review team at the National Center for Health Statistics also helped, by reaching out to WHO to check what might be the best placement.
We understood that the placement doesn’t matter so much to us as patients because all you need is a code and what that code will give you access to (and easy of tracking). But we have seen for other diseases how disagreements over the coding section meant they had to resubmit, and then resubmit again, and that means 6 more months each time since there are only two review opportunities a year, so it is important to get the proposed placement right.
We ended up applying for a code within the overall epilepsies classifications following the WHO recommendation, although CDD could fit equally well in multiple places. During all of this feedback collection process I kept getting back to the medical societies to make sure they agreed with the proposed code and were fully informed about why one code proposal and not other.
STEP 2 – FACE TO FACE REVIEW
After our application we got the news that we had been invited to the review, so we were into the agenda. That was already good news.
We then had to send a clinician to the public review at the CDC, which took place September 11, to explain in a couple of minutes why we needed a code and why THAT code in particular (these are professional coders, remember).
We sent them a very charming young neurologist who runs one of the CDKL5 Centers of Excellence. It is hard to say no to Dr Eric Marsh, and he did a great job at answering the questions after his presentation (starting by “why an epilepsy code and not a neurological or genetic code?”).
There is an online link to follow the reviews remotely. I watched it over dinner (European time) with my family and I felt we were watching the Academy Awards.
Except you still don’t know if you got the award that night.
STEP 3 – SUPPORT LETTERS
After the face to face review there is an open consultation review of about 2 months, during which the code users (for example hospitals) can review all what was presented during the September review and reach out to the National Center for Health Statistics agreeing or disagreeing with the proposals. We were encouraged to request support letters to be sent to the review team during this period.
We got letters from some of the major medical societies (others don’t produce letters but call and talk to the review team, so we still wanted to make sure they would be supportive), from relevant umbrella patient organizations like the Epilepsy Foundation and NORD, and from all of the pharmaceutical companies working on CDD.
The message was always the same: “CDD is a unique disorder with unique medical needs that therefore needs a unique code, and we support the proposed code”.
We had already told all of them since before the June application deadline that we will be applying, discussed the specific code with the medical ones, sent the face to face agenda and on-line link to all of them for them to be aware of the September 11 review, and then finally collected their letters. Between Heidi Grabenstatter from IFCR and I we sent and received over 100 emails back and forth with all these organizations for the ICD-10 proposal.
Two months after closing that consultation period, last week we got the email we were waiting for: code accepted, will be included in the next revision of October 1st.
We have now prepared a press release and will make sure we notify all families and their clinicians so they can start incorporating the new code into their medical records as soon as it is possible.
WHAT WAS KEY
I think we had two key arguments that helped us get a code created for CDD. This means that if your rare disease is at an earlier stage of medical literature and therapy development you might not get it.
1 – There were some very strong medical literature articles stating in very conclusive terms that CDD is a unique disorder, not under Lennox-Gastaut syndrome (which had a code) and not under Rett syndrome (also with a code) and not under any other disease, just a standalone disease.
2 – We have 4 drugs in clinical trials. The best way to justify that you not only have a unique disorder, but that is also has unique medical needs, is to show that there are drugs in development specifically for CDKL5 deficiency disorder. This means that companies and regulatory agencies agree you need unique drugs! Quite a strong argument. Here is where I think many other rare diseases might face difficulties.
THE SUPPORT
I believe there are very few people in patient communities who will go through the process of a requesting an ICD-10 code creation more than once. I have only had to do it for CDD, and same for my counterpart Heidi at IFCR. So we relied on a lot of advice.
We needed people who has done it before not only for specific feedback on our applications materials, but even to have a mental picture of the whole process. How longs will it take? Is that hard? Who should we talk to? Is it normal is the application team doesn’t reply to emails?
Luckily for us two friend patient groups (Angelman and Dravet) had recently gone through the process, so we had extremely useful advice from Terry Jo, Nicole and Mary Anne. The team at Ovid Therapeutics also gave us advice, since they had supported those two previous applications.
And not just advice, but we got to see their application letters. That was fantastic.
We also had the support from the US network of CDKL5 Centers of Excellence, all of which not only reviewed by also co-signed the application. When you have some many hospitals backing up a proposal, you know the reviewers have to take it seriously. And Dr Marsh was willing to free up a whole day with very short notice (think 4 business days before the face to face review in Maryland). He and I worked through the night to put together the presentation, which has specific requirements down to how to name a file without using spaces…. so believe me that was quite some challenge.
And last, the support letters. I feel fortunate that so many medical organizations, large patient organizations and pharm companies would be so willing to support us in our request for an ICD-10 code. This includes people who we didn’t know before we started this process, and who would make sure the Board of their society will have the perfect letter for them to sign before deadlines. Huge thank you to all of you.
Because we had so much help and support I would like to offer the same to those of you starting this journey: if you are a rare disease patient group and are considering applying for an ICD-10 code for your disease, reach out to me and I will be happy to be for you what all of these people were for us.
I hope this entry was informative and useful. Let me know if you have questions!
Ana Mingorance, PhD

¿Quien puede entrar en un ensayo clínico?
Cada vez que se anuncia un nuevo ensayo clínico los pacientes y sus familias experimentan una mezcla de emociones. De un lado la alegría y esperanza de saber que vienen nuevos fármacos para su enfermedad. Del otro lado, muchas veces, desilusión y sensación de injusticia al ver que muy posiblemente no tengan la oportunidad de participar en el ensayo. Como científico involucrado en el diseño y ejecución de ensayos clínicos quiero compartir con los pacientes y sus familias algunas notas sobre por qué no todos los pacientes pueden entrar en un ensayo clínico, y si esto es “justo” o “injusto”.
Cada vez que se anuncia un nuevo ensayo clínico los pacientes y sus familias experimentan una mezcla de emociones. De un lado la alegría y esperanza de saber que vienen nuevos fármacos para su enfermedad. Del otro lado, muchas veces, desilusión y sensación de injusticia al ver que muy posiblemente no tengan la oportunidad de participar en el ensayo.
Como científico involucrado en el diseño y ejecución de ensayos clínicos quiero compartir con los pacientes y sus familias algunas notas sobre por qué no todos los pacientes pueden entrar en un ensayo clínico, y si esto es “justo” o “injusto”.
NO TODOS LOS PACIENTES CUALIFICAN
1 - UN ENSAYO CLÍNICO ES UN EXPERIMENTO MÉDICO
Para los que desarrollan los fármacos y para las agencias reguladoras, un ensayo clínico es un experimento. Hay un fármaco experimental con potencial para tratar una enfermedad, pero no hay ni garantías de que funcione ni garantías de que sea seguro. Eso solo se sabe tras realizar los ensayos.
De momento en el síndrome de Dravet y de deficiencia en CDKL5 y otros estamos teniendo suerte, los ensayos clínicos están funcionando y dando buen perfil de seguridad. Pero no es engañéis, esto es solo suerte y buena ciencia. Preguntad a alguien de la comunidad del síndrome de X Frágil, o de Duchenne. Hay muchos ensayos que fracasan, e incluso algunos (pocos) que hay que parar por problemas de seguridad.
Por eso los ensayos están pensados para proteger a los pacientes y no exponer a más de los absolutamente necesarios a un posible riesgo. Tras los primeros ensayos ya se abre la puerta a muchos mas pacientes, pero sobretodo al principio la cosa tiene que ser mucho mas controlada y diseñada con la mente fría.
¿Sabíais que uno de los primeros voluntarios para una terapia génica era un muchacho con 18 años que se ofreció voluntario al ensayo y murió 4 días después por culpa de la terapia? Los ensayos clínicos son experimentos, por eso hay tanto control y restricciones en torno a los ensayos.
2 – EL EXPERIMENTO CONDICIONA LAS EDADES
Al necesitar extraer conclusiones científicas, se suele diseñar el primer ensayo de un fármaco en torno a las edades ideales para el tratamiento. Eso no quiere decir que pacientes de otras edades no se pudieran beneficiar del fármaco, o que “no lo merezcan”, pero no se trata de eso, se trata de obtener una respuesta científica con el menor número de pacientes posibles y en el menor tiempo posible. Cuanto antes se confirme si el fármaco es efectivo y seguro antes se aprueba y llega a todo el mundo.
En general se evitan edades menores de 2 años (a veces menores de 4 años) porque los niños pequeños metabolizan los fármacos de manera diferente y es mejor esperar a ver la seguridad en mayores antes de exponerles a un posible riesgo. En los síndromes con epilepsia se suele también poner un techo de edad, de 18 o 20 años, porque la mayoría de los pacientes diagnosticados son de edades inferiores y se busca crear un grupo de ensayo “representativo”.
Por eso los muy pequeños o los mayores de edad se suelen quedar fueras de los ensayos, sobretodo si es el primer ensayo con ese fármaco.
3 – TAMBIÉN CONDICIONA EL TIPO DE SÍNTOMA QUE SE PIDE
A veces los fármacos van destinados a tratar un síntoma en concreto. Por ejemplo “fenfluramina para el tratamiento de crisis epilépticas en el síndrome de Dravet”, o “ganaxolona para el tratamiento de crisis epilépticas en el síndrome de deficiencia en CDKL5”. Fijaros que no hablan de tratar la enfermedad en sí, sino su epilepsia. En ese caso un criterio para cualificar para el ensayo es tener ese síntoma con suficiente gravedad como para poder ser evaluado durante el ensayo clínico. Eso impone un mínimo de crisis epilépticas al mes, en estos ejemplos, y deja fuera a los que están sin crisis o que tienen muy pocas al mes.
No se trata de injusticia, sino de que la terapia trata un síntoma y si no se tiene (o si es demasiado infrecuente) no hay manera de saber si la terapia está funcionando.
4 – TAMBIEN CONDICIONA (A VECES) EL TIPO DE MUTACIÓN QUE SE PIDE
Además de tener el diagnóstico y la edad y síntomas apropiados, algunos fármacos requieren un tipo de mutación en concreto por la forma en que funcionan. Por ejemplo el ensayo de ataluren en Dravet y CDKL5 requería tener una mutación non-sense en el gen SCN1A o el gen CDKL5. No valen otro tipo de mutaciones, incluidas las de cambio de pauta de lectura (frameshift) aunque esas produzcan una non-sense secundaria.
Eso es así porque solo se aceptan en el estudio aquellos pacientes que tienen el problema genético que el fármaco está diseñado para tratar.
La justificación de este criterio es evidente, pero se pueden dar escenarios que son más difíciles de explicar a los padres. Por ejemplo en el caso de Stoke su terapia va a aumentar los niveles de expresión de las dos copias de SCN1A, la buena y la mala. Para eso tenemos que estar 100% seguros de que la copia mala da lugar a que se pierda la producción (o función) del canal de sodio que ese gen codifica. Si no estamos seguros de que la mutación es de pérdida de función, y queda la duda de que pudiera dar lugar a un canal que funciona mal pero por exceso de función (con alguna mis-sense) lo mejor es no arriesgarse a hacer las cosas aún peor con la terapia y estos pacientes no se deben admitir al estudio.
5 – Y DE FORMA IMPORTANTE, CONDICIONA DONDE SE REALIZA EL ENSAYO
El objetivo del primer ensayo clínico en pacientes con encefalopatías epilépticas es ver si tiene eficacia y sobretodo si es seguro. Hasta ese ensayo los experimentos son en animales, siempre hay un riesgo. Por eso el primer ensayo empieza siempre pequeño (suele ser de 20 pacientes o menos) y muy controlado. Por esta razón se hace solo en Estados Unidos donde hay más pacientes en solo un país.
Una vez ese ensayo concluye y se ve que el fármaco es efectivo y seguro, entonces ya se hacen ensayos grandes de 100 o más pacientes y se abre a más países. Porque en ese momento ya hay menos dudas sobre la seguridad y más claridad de que parece tener eficacia. El objetivo de esos ensayos grandes es CONFIRMAR estos hallazgos, y obtener datos sólidos para poder pedir la aprobación.
Por eso es normal ver que los primeros ensayos se quedan todos en América.
INCLUSO SI CUALIFICAN, NO TODOS PUEDEN ENTRAR
Otro tema importante es que una vez definido el protocolo del ensayo, la oportunidad no llega a todos los afectados del mundo que cumplan esas condiciones, ni siquiera si están dispuestos a viajar a estados unidos.
6 – NO TODOS SE ENTERAN DE QUE HAY UN ENSAYO
No todo el mundo sabe que hay ensayos clínicos para su enfermedad o la de su hijo, y no todos los médicos están al corriente tampoco. El informar a la comunidad de pacientes es una de las funciones principales de los grupos de pacientes, y aunque solo fuera por esto vale la pena estar conectados.
7 – ENTRA SOBRETODO EL QUE TIENE MÁS POSIBILIDADES DE IR AL HOSPITAL QUE TIENE EL ENSAYO
Esto es evidente. Si el ensayo está solo en 5 ciudades grandes americanas, los pacientes de la zona tienen muchas más posibilidades de entrar al ensayo porque posiblemente su médico se lo proponga. Hay familias que pueden permitirse viajar desde Asia o Europa o Latinoamérica a estados unidos, pero serán pocas y no siempre encontrarán plazas. En general, la cercanía a un ensayo clínico determina mucho las posibilidades de acceso porque ofrece muchas facilidades a los locales.
8 – ENTRA EL QUE LLEGA PRONTO
Como los ensayos tienen plazas limitadas, sobretodo los primeros ensayos en pacientes que son pequeños, es posible quedarse fuera del ensayo porque se cierra el reclutamiento cuando algunos pacientes todavía están siendo valorados.
En general hay que empezar a tener las discusiones con el médico antes de que empiece el ensayo, para no arriesgarse a quedarse fuera por plazos.
9 – ENTRA EL QUE NO SE TOPA CON RETRASTOS FUERA DE SU CONTROL
Hay casos tristes en los que por un retraso burocrático (por ejemplo en la agencia española del medicamento) el reclutamiento en ese país tarda mas y la empresa llena el cupo con pacientes de otros países. Esto es un caso real.
Esto es así porque las empresas no asignan un cupo fijo de plazas a cada hospital. Si necesitan 50 pacientes, por ejemplo, abren el reclutamiento en una treintena de hospitales en varios países y en cuando se llega 50 pacientes que cualifican para el ensayo se cierra el cupo. Y puede darse el caso de que un centro tenga 8 niños en un ensayo y otros, por lentitud del centro o su agencia reguladora, se queda a cero.
10 – ENTRA NO QUIEN DECIDE EL MÉDICO, SINO AL QUE LA EMPRESA DA EL VISTO BUENO FINAL
Y luego hay detalles que se entienden si se sabe como funciona un ensayo pero que pueden parecer completamente arbitrarios desde el lado del paciente.
Imaginad un niño con el diagnóstico que toca, la edad ideal de entre 2 y 18 años, la mutación correcta, muchas crisis al mes, en un hospital que ya está apuntando a otros pacientes al ensayo, el ensayo todavía busca pacientes y su médico le da el visto bueno y le registra para el ensayo. Y les llega un comunicado de la empresa que dice que no le pueden aceptar porque han reclutado demasiados adolescentes ya, y las agencias reguladoras les piden que equilibren los mayores y menores de 6 años para asegurarse de que el fármaco no funciona solo en los mayores o solo en los mas pequeños. Así que los últimos que entren al ensayo tienen que ser todos menores de 6 años para no desequilibrar el grupo.
Y es que el médico verifica que el paciente cumple las condiciones y lo propone al coordinador del ensayo, pero al final es este representante de la empresa (siguiendo indicaciones de las agencias reguladoras) el que acepta o no al paciente en concreto en el ensayo.
ALTERNATIVAS DE ACCESO
Yo entiendo perfectamente que para el paciente y sus familiares, e incluso para muchos médicos, un ensayo es una oportunidad para probar un fármaco que quizás sea por fin el que funcione en ese paciente. Por eso las decepciones y sensación de injusticia cuando un paciente, queriendo entrar en el ensayo, se queda fuera.
Cuando una terapia está empezando el primer ensayo clínico, lo habitual es que la única manera de tener acceso sea entrando en ese ensayo clínico, que os recuerdo que es un experimento científico por lo que acepta pocos pacientes y con unos requisitos muy específicos. No hay garantía de que funcione, y no hay garantía de que sea seguro. Pero una vez ese ensayo ha terminado y se tiene más confianza en la seguridad del fármaco se abren otras oportunidades de acceder al fármaco experimental.
En ese momento se abren opciones como el acceso por vía compasiva, que no es siempre una posibilidad pero en terapias que están ya en ensayos finales se suele ofrecer a un número limitado de pacientes. Al contrario que en el ensayo clínico, en este caso las empresas si suelen alocar un número de plazas a un médico en concreto, para usar baso su criterio en un grupo de pacientes que en general no hubiera podido entrar al ensayo clínico. Por ejemplo por ser mayores de edad, o por tener muy pocas crisis epilépticas. Esto permite a la empresa tener mas datos sobre la seguridad del fármaco, y al médico averiguar si hay eficacia más allá de en el grupo de pacientes que cumple los requisitos el ensayo. Se suele dar a los médicos más relevantes, que son responsables del ensayo clínico en ese país y que por tanto tienen experiencia de primera mano con el fármaco. Así que también es recomendable que además de tener vuestro especialista habitual estéis en contacto con alguno de los hospitales que suelen tener ensayos clínicos, porque estos son los centros que posiblemente consigan acceso a fármacos por vía compasiva.
Tras el primer ensayo clínico en pacientes, generalmente solo en Estados Unidos, se abre también la vía de entrar en los ensayos finales (ensayos pivotales) que si que suelen llegar a mas países. A veces la solución es esperar un año más para la posible llegada del ensayo a vuestros países. Este es el caso sobretodo de Europa, y dentro de Europa sobre todo de Europa occidental, pero para algunos países como en Latinoamérica los ensayos clínicos en enfermedades raras muy posiblemente no lleguen. En este caso generalmente toca esperar a la aprobación final del fármaco, o cambiar de país.
Y una vez el fármaco resulta aprobado para una enfermedad o mutación genética en concreto, se suele aprobar para todas las edades mayores de 2 años (menores no, inicialmente, por diferencia en metabolismo de fármacos) independientemente de el grado de afectación de los pacientes. Pero entre el comienzo del primer ensayo clínico y la aprobación del fármaco suelen pasar unos 4 años.
Espero no resultar muy deprimente con estas explicaciones. Suelo ver muchas preguntas al respecto cada vez que se anuncia un nuevo ensayo clínico y quiero asegurarme de tener estas explicaciones escritas para poder referenciar esta página a los pacientes o familiares que tienen dudas sobre quien puede entrar en un ensayo clínico (en concreto en enfermedades raras).
En definitiva espero que os sea de utilidad, y decidme si quedan preguntas que no he cubierto en el texto.
Ana Mingorance PhD

Repaso del congreso de la Sociedad Americana de Epilepsia (AES 2019)
El congreso de la Sociedad Americana de Epilepsia (AES por sus siglas en ingés) es la mayor cita en epilepsia del año, y como tiene lugar cada mes de Diciembre sirve también para hacer un repaso de como ha avanzado el campo a lo largo del ultimo año. AES 2019 fue el año de las terapias genéticas para las encefalopatías epilépticas y de desarrollo. Este artículo es el resumen de lo que me pareció mas interesante del congreso AES 2019
El congreso de la Sociedad Americana de Epilepsia (AES por sus siglas en inglés) es la mayor cita en epilepsia del año, y como tiene lugar cada mes de Diciembre sirve también para hacer un repaso de como ha avanzado el campo a lo largo del ultimo año.
Yo me dedico a buscar terapias para los síndromes genéticos con epilepsia, y en los últimos años estos han dominado gran parte de la agenda del congreso de AES.
Hace dos años, en el congreso de 2017, ya se veía el gran interés en las epilepsias raras (las que afectan a menos de 5 de cada 10.000 nacimientos), y todo el mundo estaba ilusionado con los resultados de cannabidiol y fenfluramina. También era evidente que hay demasiados síndromes genéticos como para intentar identificarlos uno a uno, y que hace falta pedir un test genético desde que los niños empiezan con epilepsia. Pero yo echaba en falta que hubiera algo más de conexión entre esos dos mundos, entre los tratamientos y la genética. Que más allá de hablar de diagnóstico genético habláramos de terapias genéticas.
Y ese gran salto llegó en 2018. Cuando escribí el resumen del congreso de AES de 2018 destaqué:
Si hay un progreso visible en AES 2018 que marca un antes y un después en el campo de la epilepsia es la llegada de los tratamientos no farmacológicos para epilepsia.
Y ese “antes y después” ha quedado aún mas claro este año. Durante AES 2019 las sesiones en las que se trató de terapias génicas y de oligonucleótidos antisentido (los ASO, que son terapias que parecen un cachito de ADN) se llenaron a capacidad completa, llenas de gente de pie, hasta el punto de que tuvieron que cerrar las puertas por nuestra seguridad. El campo de la epilepsia, hasta ahora dominado por fármacos, a abrazado de lleno la biotecnología y hemos entrado oficialmente una nueva era.
Antes de pasar al repaso del congreso, quiero clarificar dos términos que usaré en el artículo:
Encefalopatías epilépticas y de desarrollo. Hasta ahora era un lío saber como llamarlas…. epilepsias genéticas, epilepsias raras, epilepsias huérfanas… siempre añadiendo la coletilla de “que son mucho más que una epilepsia”. Ahora ya tienen nombre. Son las encefalopatías epilépticas y de desarrollo, que no son todas genéticas (por ejemplo Lennox-Gastaut) y que sin embargo si que comparten problemas de desarrollo neurológico que no es solo consecuencia de la epilepsia. El término fue acuñado por la Liga Internacional Contra la Epilepsia en 2017 y ha cuajado.
Terapias genéticas. Cuando hablamos de terapias génicas nos referimos a aquellas que usan genes como terapia, como el típico virus que aporta una copia del gen humano sano. Pero está claro que nuestro armamentario terapéutico contempla muchas otras maneras de tratar la causa genética de enfermedades. El Doctor Barry Ticho, de Stoke, usó la expresión “terapias genéticas” para referirse a su terapia que es un oligonucleótido, porque es para tratar el problema genético (aunque la terapia no sea un gen). Y me pareció un término fantástico, así que lo usaré para referirme a las terapias que actúan sobre los genes o el ARN, con independencia de que estas usen virus, oligos u otras modalidades.
LAS 5 LECCIONES DEL CONGRESO AMERICANO DE EPILEPSIA
En una frase: AES 2019 fue el año de las terapias genéticas para las encefalopatías epilépticas y de desarrollo.
En más detalle, esta es la lista de lo que me pareció mas interesante del congreso AES 2019:
1. MÁS ALLÁ DE LOS SÍNTOMAS: TERAPIAS GENÉTICAS
En los últimos años hubo varios grandes proyectos internacionales sobre las bases genéticas de la epilepsia que identificaron cientos de genes que causan las epilepsias epilépticas y de desarrollo además de otras formas de epilepsia.
La empresa Stoke tiene una diapositiva en su presentación a inversores con cifras que me resultan muy impresionantes. Dice:
· 50 millones de persona en el mundo viven con epilepsia
· El 50% de las epilepsias tienen una causa genética identificada
· Hay más de 180 genes implicados
· Hay 0 terapias contra estos genes
Su programa para el síndrome de Dravet es uno de los más avanzados que buscan cambiar ese número cero. Es increíble que con cientos de causas genéticas, que causan síndromes con problemas severos de neurodesarrollo además de epilepsia resistente a terapias, no tengamos hasta ahora ni un solo tratamiento para esos genes o para sus consecuencias directas. Porque no hay que ir tan lejos, para muchos de estos síndromes debería ser posible desarrollar fármacos de los convencionales que puedan corregir el problema en la proteína que es responsable de su enfermedad. Como en la fibrosis quística, que hay varios fármacos para corregir el canal CFTR. Y varias de estas estrategias ya están en marcha también para algunas epilepsias.
Si repasamos tan solo los programas desarrollados por empresas (no los académicos), y a sabiendas de que se me escapan algunos, esta es la lista de los tratamientos de este tipo que hay actualmente en desarrollo (gracias Steve Petrou por resumir muchos de ellos en tus presentaciones!):
Síndrome de Dravet causado por mutaciones en SCN1A: aumento de expresión de SCN1A por Stoke (ASO) y Encoded Therapeutics (virus), y en fases más tempranas activadores del canal de sodio por Lundbeck y Xenon (fármacos)
Desordenes asociados a SCN2A: reducción de expresión de SCN2A por RogCon/Ionis (ASO) e inhibidores del canal por Xenon y Praxis (fármacos)
Encefalopatía epiléptica y de desarrollo causada por SCN8A: inhibidor del canal por Xenon (fármaco)
Encefalopatía epiléptica causada por KCNQ2: moduladores del canal por Xenon y Knopp (fármacos)
Encefalopatía epiléptica causada por KCNT1: reducción de expresión de KCNT1 de Ionis (ASO)
Síndrome por deficiencia en CDKL5: terapia de reemplazo enzimático por Amicus y terapia génica por Amicus y Ultragenyx (virus)
Síndrome de Angelman: activación de la copia paterna de UBE3A de Ionis, Roche y Ultragenyx (ASO), terapia génica por PTC y Pfizer (virus) – ¡en total 5 programas!
Y la lista no está completa, estos son solo los programas que fueron presentados en AES y algunos que conozco. Además vimos en AES experimentos preliminares de que la plataforma de Stoke puede también aumentar los niveles de expresión de SynGAP1, con lo que está claro que estas tecnologías llegarán a mas encefalopatías epilépticas y de desarrollo. Y luego están todos los programas académicos que podrían también llegar a ensayos clínicos en los próximos años.
Esto es a penas el comienzo.
2. MÁS ALLÁ DE LAS CRISIS EPILÉPTICAS
Otra cosas que me ha gustado mucho es empezar a oír a los neurólogos diferenciar lo que es epilepsia (la enfermedad) y lo que son las crisis epilépticas (un síntoma).
Hasta hace poco hablaban de “epilepsia” para referirse solo a las crisis, y usar el término “comorbilidades de la epilepsia” para los aspectos que no son crisis epilépticas y que suelen venir asociados con la enfermedad, por ejemplo la depresión, el retraso cognitivo, o los problemas motores. Llamarlos comórbidos apuntaba a un papel secundario, menos importante.
En los últimos dos años oigo mucho más a los médicos hablar de “epilepsia” como inclusivo de ambos, crisis y otros aspectos. Expandiendo el concepto de epilepsia para ir más allá de las crisis abre la puerta a desarrollar terapias que también van más allá de las crisis. Y vemos mucha más investigación en estos otros aspectos de la enfermedad, que en el caso de las encefalopatías epilépticas y de desarrollo son un aspecto central de la enfermedad. Y se empieza a hablar de fármacos anti-crisis (anti-seizure) para referirse a los fármacos que usamos hasta ahora en epilepsia y que solo tratan las crisis, en vez de llamarles fármacos antiepilépticos (porque no tratan la enfermedad). Estos no son solo cambios lingüísticos, son una ampliación de lo que incluye la especialidad médica de la epilepsia, y un gran paso adelante para los pacientes.
3. ¿Y AHORA QUÉ? LA DIFICULTAD DE DISEÑAR ENSAYOS CLÍNICOS QUE VAN MÁS ALLÁ DE LAS CRISIS
La buena noticia es que ahora sabemos que la epilepsia implica mucho más que crisis epilépticas y empezamos a tener terapias que podrían también tratar estos aspectos de la enfermedad.
La mala noticia es que no sabemos como diseñar esos ensayos clínicos que van “más allá de las crisis”, o qué quieren ver las agencias reguladoras de estos fármacos.
Actualmente hay muchísima actividad en este tema. Vimos presentaciones en AES 2019 de empresas y de académicos sobre qué medir en ensayos clínicos en algunos de estos síndromes, y cada vez que hablo con grupos de pacientes para aprender de sus ensayos clínicos tienen la dificultad del diseño de los ensayos como primer punto de la lista: cómo diseñar ensayos clínicos que capturen mejorías más allá de reducción de crisis, o cómo diseñar ensayos que muestren cambios en el curso de la enfermedad.
No tenemos las respuestas, es work in progress, pero andamos buscando la respuesta todos juntos y me alegré de ver que el desafío de diseñar estos ensayos clínicos era tema de discusión en AES.
4. ABRIENDO LA COMUNIDAD
Todo el que ha ido a un congreso de AES sabe que hay una población de pacientes muy activa. Y esto pasa porque las encefalopatías epilépticas y de desarrollo son enfermedades raras, y las enfermedades raras son notables por movilizas la comunidad de pacientes (o padres de personas afectadas). Una consecuencia es que durante el congreso de AES hay cada vez más reuniones satélite organizadas por los grupos de pacientes centradas en su síndrome, y vemos a los representantes de pacientes en la sala de exposiciones y las salas de conferencias, participando en la conversación y hablando con empresas.
Todo esto está muy bien, pero quiero poner sobre la mesa dos ideas para hacerlo mejor, ya que los grupos de pacientes son tan centrales en la comunidad de epilepsia:
1. Tenemos que hacer más fácil la participación de grupos de pacientes en AES. Yo he ido a muchos congresos representando a grupos de pacientes, y los gastos son demasiado altos cuando se trata de ahorros personales o fondos de una ONG. Eso nos lleva a ver situaciones como solo pagar por la matrícula del congreso de algunos participantes, y pasarse la acreditación de uno a otro para entrar a las salas. O a simplemente no registrar a nadie y simplemente enlazar reuniones con médicos y empresas una de tras de otra en el lobby del hotel más cercano. Esto lo sabemos todos, y no debemos ignorarlo. Propongo ayudar a la participación de representantes de pacientes en los congresos médicos de su campo ofreciéndoles matrícula gratuita (como mínimo) y posiblemente con una bolsa de viaje, que podría ser financiado por empresas y gestionado por la organización del congreso. A mi no me hace falta ver pegatinas en el suelo del hotel anunciando (y pagado por) una empresa farmacéutica; lo que quiero ver es a los grupos de pacientes ser invitados (y ayudados) a estar en la sala como miembro esencial de la comunidad.
2. Tenemos que hacer más fácil la organización de simposios y mesas redondas organizadas por pacientes durante el congreso. Ahora mismo estas no son parte oficial del congreso de AES, con lo que los grupos de pacientes intentan hacer que sus reuniones sean convenientes buscando la tarde del día anterior o el horario de desayuno y reservando salas en hoteles colindantes al congreso. Y a menudo caen varios a la vez, porque no hay tantos huecos, con lo que los profsionales que trabajan en varios de esos síndromes tienen que elegir a cual ir y a cual no. Propongo crear un tiempo protegido durante el congreso para poder tener estas reuniones. Por ejemplo ya es tradicional que la Fundación Síndrome de Dravet americana organice su mesa redonda la tarde de la víspera del congreso, con lo que los que trabajamos en Dravet sabemos que tenemos que llegar al congreso un día antes. ¡Y funciona muy bien! Con lo que podemos imaginar un programa en el que el primer día es dedicado a estas reuniones organizadas por pacientes, y luego empiezan los 4 o 5 días de agenda médica. Lo que no podemos hacer es seguir ignorando el hecho de que las organizaciones de paciente son no solo participantes del congreso, además generan parte de la agenda – hasta ahora de forma no oficial. Y esta es la realidad, con lo que debemos incorporarlos en la agenda oficial en la medida de lo posible,
Me gustaría saber qué pensáis de estas propuestas.
5. HAY UNA GRAN URGENICIA POR HACER TESTS GENÉTICOS
Lo de hacerle un panel genético a una persona con epilepsia ya no es “algo interesante”, es algo necesario. Con el descubrimiento de cientos de genes y ahora con la llegada de las terapias genéticas tenemos que saber identificar a todos estos pacientes (además de otros aspectos valiosos de saber la causa de tu enfermedad). BioMarin, Stoke y Xenon patrocinan un programa de test genético gratuito (bueno, pagado por ellos) a través de Invitae, que hasta ahora incluye a todos los niños en EEUU y Canadá con 5 o menos años de vida y epilepsia, y ahora se abrirá hasta los 8 años.
Este es un modelo ejemplar y posiblemente la forma más efectiva de llegar a todos los niños con epilepsia. Así que agradezco y doy la enhorabuena a las 4 empresas por ofrecer este programa.
El paso siguiente que me gustaría ver es la expansión de este programa más allá de América del norte (posiblemente a través de otros laboratorios genéticos) y ampliar las edades. Espero que los datos que está obteniendo Invitae sirvan para documentar el beneficio de estos tests genéticos patrocinados, y que lleve por tanto a su globalización.
MIRANDO A 2020
Estoy deseando ver la llegada de los ensayos clínicos con terapias genéticas en 2020. Desde los fármacos contra mutaciones de canales iónicos específicas a la llegada del primer tratamiento genético para el síndrome de Dravet a ensayos clínicos.
El NINDS (la rama de neurología del Instituto de Salud Norteamericano) define sus propiedades para investigación en epilepsia cada 7 años. La última reunión fue en 2013, de donde salieron las prioridades de investigación de 2014. Y en abril de 2020 vuelve esa reunión, llamada “Curando las epilepsias 2020”, de donde saldrán las prioridades de investigación para los próximos 7 años. Para mi es un honor ser uno de los representantes de la Sociedad Americana de Epilepsia (si, tienen miembros internacionales) en la comisión conjunta entre la Sociedad y el NINDS que está trabajando en esas prioridades y en la conferencia de Curando las epilepsias 2020. Y como la investigación en general también es internacional, os animo a los investigadores a enviar vuestras propuestas de nuevas prioridades a través de este enlace hasta el 20 de febrero.
Y también estoy deseando ver durante 2020 más noticias de otros síndromes, porque se que hay muchos progresos que no han sido presentados en AES (como mucho de lo que pasa en CDKL5) y porque otras enfermedades tendrán grandes avances durante el año. Espero poder ver todos estos progresos el año que viene.
Porque está claro que sobre todo, el 2020 será el año de los ensayos clínicos con terapias genéticas en encefalopatías epilépticas y de desarrollo. Va a ser un gran año.
Ana Mingorance, PhD

Top 5 insights from the American Epilepsy Society meeting (2019)
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. AES 2019 was the year of genetic therapies for the developmental and epileptic encephalopathies. This article highlights what I found the most interesting at the AES 2019 meeting.
Versión en español en este enlace
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. I look forward every year to the first week of December for this reason.
I look for therapies for rare genetic epilepsies, and in the recent years this area has exploded to take over much of the AES meeting.
Two years ago, there was already growing attention for the orphan epilepsies* and everybody was excited about cannabidiol and fenfluramine. It was starting to become clear that there are just too many genetic syndromes to try to diagnose them one by one, so we need to run genetic epilepsy panels as soon as possible in children. AES 2017 was a great meeting, but I was still hoping there would be more of a connection between treatments and genetics. That we could move beyond genetic diagnosis and towards genetic treatments.
And we made a big jump in that direction at the American Epilepsy Society meeting in 2018. I wrote in that article:
There was one key progress visible at the AES 2018 meeting that defines a before and after moment in the field of epilepsy, and this is the arrival of non-pharmacological therapies for treating epilepsy.
That before and after was tremendously clear during this year’s conference. During AES 2019, the sessions on genetic therapies and antisense oligonucleotide (ASO) therapies for rare epilepsies were filled to full (standing) capacity and the organizers had to close the doors. The epilepsy field, which has been until recently dominated by pharmacology, has embraced the technologies of biotech and we have officially entered a new era.
Before I review the main take-home messages from the meeting, I want to clarify two terms that I will be using:
Developmental and epileptic encephalopathies. We don’t need to struggle with calling them “genetic epilepsies” “rare epilepsies” or “orphan epilepsies” anymore, always adding the message that “they involve much more than just epilepsy”. Now the field has a name. We are talking about the developmental and epileptic encephalopathies (DEEs), where not all are genetic (hello LGS!) but they all have a strong neurodevelopmental component that is not 100% due to the epilepsy. The term was introduced by the ILAE in 2017 and it is staying.
Genetic therapies. When we talk about gene therapies, we talk about using genes as a therapy (as in the classical AAV carrying a healthy copy of a human gene). But it is clear that our therapeutic armamentarium to target genetic diseases goes beyond that. Dr Barry Ticho from Stoke referred to their oligonucleotide therapy as a “genetic therapy”, because it treats the genetic problem even though the therapeutic agent does not deliver a gene. I loved the term, so I will use it as well to incorporate all the different DNA and RNA-targeting therapeutics regardless of them using virus, ASOs or other modalities.
MY TOP 5 INSIGHTS FROM THE AMERICAN EPILEPSY SOCIETY MEETING
In one line: AES 2019 was the year of genetic therapies for the developmental and epileptic encephalopathies.
In more detail, here is the list of what I found the most interesting at the AES 2019 meeting:
1. BEYOND SYMPTOMS: GENETIC THERAPIES
In the recent years, many large efforts in understanding the genetic basis of epilepsies led to the identification of hundreds of genes causing developmental and epileptic encephalopathies as well as other epilepsies.
Stoke Therapeutics has a slide in their investors presentation with very impressive numbers. It says:
· 50 million people globally affected by epilepsy
· 50% of epilepsies have an identified genetic cause
· More than 180 disease-associated genes
· 0 genetically-targeted therapies for epilepsies
Their Dravet syndrome program is one of the most advanced therapies that will hopefully change that number zero. It is unbelievable that with hundreds of genetic causes, often leading to severe neurodevelopmental syndromes with aggressive epilepsy and high unmet need, we have so far not one single therapy that corrects the genetic defect or its direct consequences. And we don’t need to get to the extreme of correcting genes, for some of these epilepsies it should be possible to develop small molecules that will target the disease-causing protein, either with activators or with inhibitors. Cystic Fibrosis has shown us how to do this with CFTR, and there are currently several small molecule approaches in development for the epilepsies aiming to correct the protein dysfunction that results from genetic mutations.
Looking only at corporate programs (not even including academic efforts), and likely missing some projects, the following treatments are in development (thank you to Steve Petrou for summarizing many in his slides!):
Dravet syndrome caused by SCN1A mutations: SCN1A upregulation from Stoke (ASO) and Encoded Therapeutics (AAV), and early stage ion channel activators from Xenon and Lundbeck (small molecules)
SCN2A-associated conditions: SCN2A downregulation from RogCon/Ionis (ASO), ion channel inhibitor from Xenon and Praxis (small molecule)
SCN8A developmental epileptic encephalopathy: ion channel inhibitor from Xenon (small molecule)
KCNQ2 epileptic encephalopathy: ion channel modulator from Xenon and Knopp (small molecules)
KCNT1 epileptic encephalopathy: KCNT1 downregulation from Ionis (ASO)
CDKL5 deficiency disorder: enzyme replacement therapy from Amicus (biologic) and gene therapy from Amicus and Ultragenyx (AAV).
Angelman syndrome: activation of paternal UBE3A from Ionis, Roche and Ultragenyx (ASO), and gene therapy from PTC and Pfizer (AAV)
The list is not complete, these are just some of the programs I know about or that were presented at AES. We also saw at AES some early data on the feasibility of using the Stoke platform to upregulate SynGAP1, so it is clear that the current technologies will be able to target many more of the developmental and epileptic encephalopathies, and the significant corporate activity in the space is a very good sign. And there are also many academic programs developing genetic therapies that might lead to clinical programs in the next few years.
This is just the beginning.
2. BEYOND SEIZURES
One aspect I particularly liked was to hear more and more neurologists make a difference between the symptom (seizures) and the condition (epilepsy).
Until recently it was common use “epilepsy” as meaning just seizures, and to hear “epilepsy comorbidities” to refer to the non-seizure epilepsy-associated aspects of the disease such as depression, cognitive delay or motor problems. Calling them comorbid hinted a secondary, less important, role in the disease.
In the last two years I hear clinicians speak more about epilepsy as inclusive of both seizures and the non-seizure aspects. By expanding the definition of epilepsy to go beyond seizures, we open the door for the development of new therapies that also go beyond seizure treatment. There is more and more research around those other aspects of the disease, which are very common in the developmental and epileptic encephalopathies, and the term anti-epileptic medications, referring to the classical drugs, is being replaced by anti-seizure medications. This is not just a linguistic shift, this is a broadening of the scope of a medical and research specialty, to the benefit of patients.
3. AND NOW WHAT? THE CHALLENGE OF CLINICAL TRIALS BEYOND SEIZURES
The good news is that we now understand that epilepsy conditions go well-beyond seizures and start having the therapies that could address the different disease aspects.
The bad news is that no one really knows how to design those “beyond seizures” trials, or what regulators will want to see from those therapies.
This is right now a space of enormous activity. We saw presentations at AES 2019 from companies and academic groups on what to measure in clinical trials for some of these disorders, and whenever I sit down with a patient organization to learn about clinical trials in the space this is a the top of everybody’s list: how to design trials to show efficacy beyond seizure reduction, or to show disease-modification (changes in the course of the disease). We don’t have the answers yet, this is work in progress, but we are all figuring this out together and I was happy to see the challenge of designing these trials also being discussed at AES.
4. OPENING THE COMMUNITY
Everyone who has come to the AES meetings knows that there is a very active patient community. This is precisely because of the developmental and epileptic encephalopathies, which are rare diseases (affecting less than 200,000 people in America), and the rare disease space has a very strong patient advocacy component. As a result, there is a growing number of satellite patient-scientist meetings during the AES congress and you can see patients (often parents) walking around the exhibition hall meeting companies, participating in the discussions and even presenting science.
All of this is great, but I want to put forward two ideas to make this better, since the patient groups are an essential part of the epilepsy community:
1. We need to make it easier for patient groups to attend AES. I have gone to many conferences representing a patient group and it is just too expensive to attend when using personal or charity funding. So we see things like only registering some attendees and swapping badges at the entry, or even not registering at all and just scheduling a bunch of meetings with companies and clinicians at the lobby of the nearest hotel. We all know this. And we should not ignore it. I propose sponsoring patient groups attendance to the major medical conferences in their field, with free registration (at a minimum) and possibly travel support sponsored by companies but managed by the conference organization. I don’t need to see floor stickers on the nearest hotel from (and paid by) a pharma company; I want to see those necessary members of the community being properly invited (and supported) to be in the room instead.
2. We need to make it easier to schedule the patient-run symposiums or round tables at the meeting. Right now these are not official part of the AES meeting so the different groups try to make it convenient to attendees by scheduling their meetings early in the day, or the day before the AES conference starts, and hosting them nearby the conference center. Often they clash (there not that many early mornings!) so many professionals who work on multiple syndromes have to choose which one to attend. I propose protecting some time before the conference for hosting these patient-led meetings. We have traditionally had the Dravet Syndrome Foundation Research Roundtable the day before AES in the evening. The main AES program starts the following day in the afternoon, and many people interested in Dravet knows that they should get to the hosting city one day before so they start with the Dravet Roundtable. That works great. I could see a program where we have a first day that is devoted to patient-organized symposiums, followed by the 4-5 days of main conference agenda. We can’t keep ignoring that the patient organizations are not only an important part of the attendees, but also contribute to part of the agenda. This is already happening. We must get them into the actual agenda as much as possible.
I would love to know what you think about these.
5. GENETIC TESTING IS URGENT
Genetic testing of people with epilepsy is not a “nice to have” anymore. With the discovery of hundreds of genes, and now with the development of genetic therapies, we need to know who those patients are (in addition to the many other values of knowing the cause of your disease). BioMarin, Stoke and Xenon sponsor a free (sponsored) genetic program at Invitae that will soon be open to all children in the US and Canada 8 years and younger. Currently it is for age 5 and younger. This is an excellent model and perhaps the most effective way of making sure that children with epilepsy get early genetic testing. I thank and congratulate the for companies for offering this program.
As a next step, I would like to see this expanded beyond north America (likely through other providers) and also into broader ages. I hope the data obtained by Invitae so far will help document the return on investment for these sponsored programs, and encourage their expansion.
LOOKING INTO 2020
I am excited to see the genetic therapies trials in 2020. From ion channel modulators that might help children with devastating syndromes to the arrival of the first genetic treatment for Dravet syndrome to clinical trials.
Also, the NINDS will host a very special epilepsy meeting called Curing the Epilepsies 2020: Setting Research Priorities in April 2020. This meeting follows one in 2013, and will help establish the NINDS Benchmarks for Epilepsy Research (see here the 2014 ones). The Benchmarks help shape the research priorities from NINDS on epilepsy for the next 7 years so these meetings are extremely important. I am trilled to be a member of the joint AES-NINDS team putting together the next edition of the benchmarks, and encourage you all to send your ideas before February 20 2020 though this link. This only happens every 7 years so don’t miss the chance to shape those priorities!
And I also look forward to more progresses in more developmental and epileptic encephalopathies next year. For example, I know there is a lot more going on in CDKL5 Deficiency Disorder than what we saw at AES, and I hope there will more presented next year.
Above all, 2020 will be the year of clinical trials with genetic therapies in development and epileptic encephalopathies. And I can’t wait!
Ana Mingorance, PhD

Repaso del Foro CDKL5 2019
La quinta edición del Foro CDKL5 tuvo lugar en Boston, los días 4 y 5 de noviembre. El Foro es una reunión anual exclusivamente por invitación que organiza la Fundación Loulou y en la que científicos y miembros de la industria farmacéutica se reúnen con representantes de la comunidad de pacientes para repasar los últimos avances en el campo. Este ha sido mi tercer Foro CDKL5, y el segundo desde que me uní a la Fundación Loulou.
Este es un repaso para los grupos de pacientes de las principales novedades del Foro CDKL5 2019. [SPANISH VERSION - ALSO AVAILABLE IN ENGLISH]
La quinta edición del Foro CDKL5 tuvo lugar la semana pasada en Boston, los días 4 y 5 de noviembre. El Foro es una reunión anual que organiza la Fundación Loulou y en la que científicos y miembros de la industria farmacéutica se reúnen con representantes de la comunidad de pacientes para repasar los últimos avances en el campo. Este ha sido mi tercer Foro CDKL5, y el segundo desde que me uní a la Fundación Loulou.

Al ser la quinta edición, el Director del Foro CDKL5 y querido amigo mío Dan Lavery ofreció un repaso de lo mucho que ha cambiado el campo desde la primera edición del Foro. ¡y ha cambiado tanto que parece que hablemos de dos enfermedades diferentes!
Así que voy a tomar prestada la revisión de Dan de “entonces versus ahora” para compartir con vosotros un repaso del último Foro CDKL5 y de cuanto ha cambiado la investigación y desarrollo de terapias para la deficiencia en CDKL5.
1. DE ENFERMEDAD ULTRA-RARA A SER UNA DE LAS CAUSAS GENÉTICAS DE EPILEPSIA MAS COMUNES
Cuando se celebró el primer Foro en 2015, se pensaba que el síndrome de deficiencia en CDKL5 (CDD por sus siglas en inglés) afectaba en torno a 200 personas en todo el mundo. Pronto nos dimos cuenta de que debía haber muchos más, pero la cifra concreta de casos ha sido muy difícil de calcular.
Este año por primera vez se publicó un estudio de incidencia de CDD, y ahora sabemos que nace un niño con CDD en cada 42.400 nacimientos. Es estudio se hizo siguiendo a todos los nacidos en Escocia durante tres años, que son más de 150.000 nacimientos, y haciendo un test genético completo a todos los niños que presentaron epilepsia antes de su tercer cumpleaños. Esta metodología hace que los resultados sean muy sólidos, y apunta a que el gen CDKL5 es una de las causas genéticas más comunes de epilepsia. Así que ahora sabemos que la cifra real no es de 200 casos, sino de más de 20.000 casos en todo el mundo. ¡para nada una enfermedad ultra-rara!
La cifra que todavía no sabemos es la prevalencia, que es el número total de pacientes diagnosticados. Esto se debe a dos razones. La primera es que como los tests genéticos solo se están haciendo en los últimos años, nos faltan por diagnosticar la mayoría de los pacientes adultos. Y la segunda es que no tenemos una forma buena de localizar a todos los casos diagnosticados para poder contarlos.
Para intentar encontrar todos estos casos, desde la Fundción Loulou y el grupo de pacientes americanos IFCR pedimos este año la creación de un código ICD-10 para el síndrome. Estos son los códigos que usan los médicos cuando diagnostican un paciente, y el tener un código nos permitirá hacer estudios epidemiológicos ya que habrá una forma de identificar todos los casos. Entre tanto, una cosa está bien clara: hablamos de buscar decenas de miles de pacientes, no unos pocos cientos.
2. DE QUINASA HUÉRFANA A REGULADOR GENERAL
Si hay un área en la que el campo de CDKL5 ha avanzado de forma brutal desde 2015 es en la comprensión de qué hace CDKL5 en el cerebro. Estaba claro que se trataba de una proteína quinasa, las que activan e inactivan otras proteínas como si fueran el interruptor de la luz, pero la identidad de esas proteínas concretas que se encienden y se apagan controladas por CDKL5, y lo que estas hacen en las neuronas, eso era desconocido.

Ahora en 2019 los científicos han identificado muchas de estas proteínas controladas por CDKL5, y han desarrollado reactivos de laboratorio (anticuerpos) para poder ver donde y cuando CDKL5 está activa en el cerebro. Los científicos han averiguado también que CDKL5 controla muchas proteínas asociadas con el esqueleto neuronal (el citoesqueleto) y que es muy posible que es a través de este proceso que CDKL5 controla la presencia de ciertos receptores en la superficie de las neuronas. Como resultado de todo esto, la falta de CDKL5 lleva a que las sinapsis (las conexiones neuronales) permanezcan más inmaduras, y que tengan en su superficie receptores típicos de neuronas más inmaduras, y eso lleva a hiperexcitabilidad neuronal.
Saber todo esto tiene dos implicaciones importantes de cara a desarrollar terapias para la enfermedad. La primera es que como CDKL5 controla tantos procesos celulares, va a ser muy difícil de compensar su falta actuando sobre otras partes de la célula. Está claro que hace falta volver a poner CDKl5, reemplazando el gen o la proteína. Las buenas noticias es que este tipo de tratamiento está ya en desarrollo.
La segunda implicación importante es que lo que no hemos visto es falta de conexiones entre partes del cerebro, cambios de estructura cerebral, o muerte neuronal o cualquier otro proceso neurodegenerativo. Por lo tanto parece que CDKL5 hace falta todo el tiempo para mantener el proceso constante de formación de conexiones entre neuronas (sinapsis). Y esto indica que si podemos devolver CDKl5 al cerebro tenemos muchas posibilidades de obtener eficacia incluso en cerebros maduros. Eso no sería así si CDKL5 hiciera falta para la migración neuronal (que pasa en bebés un niños muy pequeños), o si la falta de CDKL5 llevara a muerte neuronal. En esos casos solo tendríamos una ventana temporal para actuar muy limitada. Pero basado en lo que sabemos ya de la biología de CDKL5, personalmente creo que CDD es una enfermedad candidata para terapia génica o de reemplazo enzimático, incluso en adultos.
3. DE CERO A CUATRO ENSAYOS CLÍNICOS, Y MÁS EN CAMINO
Otro aspecto que ha cambiado dramáticamente en CDD desde 2015 es el interés de la industria farmacéutica en la enfermedad. La primera empresa en tomar el testigo fue Marinus, y anunciaron este Foro que están cerca de cerrar el reclutamiento de su ensayo clínico cumpliendo los plazos planeados. Y estamos hablando de 100 pacientes.
Pensad en la cifra: en a penas un par de años hemos pasado de pensar que no hay más que 200 pacientes en todo el mundo, a ser capaz de hacer ensayos clínicos de 100 pacientes a la vez que hay otros ensayos en paralelo.
Porque Marinus no está solo, hay tres ensayos clínicos adicionales en CDD ahora mismo, todos en fase 2. PTC acaba de completar el ensayo con ataluren en pacientes con CDD debido a mutaciones non-sense y están analizando los datos. Ovid y Takeda tienen un ensayo en marcha con el fármaco TAK-935. Y la universidad de Nueva York está realizando un ensayos clínico con fenfluramina, el fármaco de Zogenix para el síndrome de Dravet. Durante el Foro, Zogenix recibió el Premio de Excelencia del Foro CDKL5 por su contribución clínica a través de este ensayo en CDD.
Así que hemos pasado de tener una enfermedad que se pensaba que era ultra-rara, y que no estaba ni reconocida como enfermedad independiente por los reguladores porque incluso la comunidad médica la confundía con Rett, a hablar de una enfermedad única claramente independiente que recibe designaciones de fármaco huérfano y que tiene cuatro ensayos clínicos por cinco empresas farmacéuticas (Takeda y Ovid trabajan juntas en su programa para CDD).
Y estas cinco empresas tampoco están solas, empresas como Amicus y Ultragenyx que estaban presentes en el Foro CDKL5 están trabajando en terapias curativas para CDD, y pudimos ver varias terapias adicionales durante el Foro:
Una colaboración entre el Trinity College de Dublín y la Universidad de Insubria nos mostró como un fármaco que modula el esqueleto neuronal (que no funciona bien cuando falta CDKL5) mejora varias de los problemas neurológicos en ratones con CDD.
Un grupo de la Universidad de Pensilvania nos enseñó como otro fármaco, que actúa solo en receptores típicos de neuronas immaduras pero que en los cerebros con CDD siguen estando presentes mucho mas tarde, también mejora varias de los problemas neurológicos en ratones con CDD.
Este mismo grupo también nos enseñó como en ratones con CDD, el cannabidiol corrige también varios problemas neurológicos, lo que cuadra con datos de GW Pharma de que Epidyolex podría tener eficacia en esta enfermedad.
E incluso la empresa Takeda nos presentó resultados con un segundo fármaco (no el que tienen en ensayos clínicos) y que también corrige algunos de los problemas neurológicos en ratones con CDD actuando sobre otras proteínas en las neuronas. Incluso han obtenido la designación de fármaco huérfano para este posible tratamiento para CDD por la agencia americana del medicamento (la FDA) hace unos meses.
Como veis cuatro ensayos en ratones que nos muestran que hay muchas más terapias en desarrollo y que podrían avanzar hacia ensayos clínicos, y que ayudarían a controlar no solo la epilepsia sino también otros de los problemas asociados con CDD. Y estos son solo algunos de los futuros tratamientos que vimos, ahora os cuento algunos más en las sesiones siguientes.
Lo que es importante es que sepáis que todas estas posibilidades nos llegan por lo mucho que hemos avanzado en nuestra comprensión de las funciones de CDKl5 en las neuronas en situaciones normales, de lo que pasa cuando la proteína falta, y de como la enfermedad se presenta en ratones cuando les falta CDKL5 (que no son los mismos síntomas exactamente que en personas). Y esto ha sido posible gracias al trabajo de los grupos de pacientes y la Fundación Loulou. Esta es una de las formas en los que los grupos de pacientes pueden tener un gran impacto, financiando los proyectos tempranos y la creación de modelos animales que su vez permiten que los grupos de investigación puedan más adelante asegurar financiación pública que suele ser tremendamente competitiva. Dar un impulso inicial para que el campo luego pueda despegar solo.
4. DE SÍNTOMAS A CURAS
Tanto los cuatro tratamientos con datos en ratones que os acabo de describir como los ensayos clínicos en marcha (salvo por ataluren), ayudan a que el cerebro funcione mejor pero ni corrigen la mutación en el gen CDKL5 ni la falta de la proteína CDKL5 en el cerebro. Ayudan a que el cerebro funcione mejor sin necesitarlos.

Tengo una diapo que uso en conferencias con familias donde explico los distintos tipos de terapias que pueden ser desarrolladas para CDD a diferentes niveles. Y la diapo es perfecta para explicar esto. Esencialmente sabemos que en CDD el cerebro no funciona bien, porque la falta la proteína CDKL5, porque tiene una mutación en el gen CDKL5. Y sabiendo esto podemos pensar en tratamientos a diferentes niveles. Por ejemplo podemos pensar en tratamientos que hagan que el cerebro funcione mejor a pesar de no tener CDKl5. O en volver a poner la proteína. O en volver a poner el gen. O en tratamientos mas complicados que puedan corregir la mutación del gen, o quizás reactivar la segunda copia del gen en el segundo cromosoma X y que no está siendo usada.
En las primeras secciones describía los progresos que han tenido lugar en el desarrollo de terapias capaces de hacer que el cerebro funcione mejor a pesar de no tener CDKL5. Pero donde hemos visto una explosión enorme en la ciencia ha sido en las otras estrategias: las que corrigen la causa de la enfermedad.

En una de las sesiones en el Foro teníamos un equipazo impresionante sobre el pódium: Kyle Fink de la Universidad de California Davis, Jim Wilson de la Universidad de Pensilvania, y David Liu del Broad Institute en Boston. Uno a uno nos enseñaron tres formas diferentes de hacer que las neuronas puedan producir CDKL5, y parecía que estuviéramos viendo la conferencia en un futuro que cada vez está mas cercano, cuando la ciencia pueda corregir las mutaciones que causan enfermedades genéticas.
Primero Jim Wilson nos enseño sus resultados con un proyecto de terapia génica que están llevando a cabo en Filadelfia y en el que están desarrollando un virus al que le quitan todo el ADN del virus y en su lugar le ponen el gen CDKL5 humano. Cuando inyectan estos virus en el cerebro de ratones con CDD, empiezan a producir CDKL5 y les consiguen mejorar muchos de los problemas neurológicos. Jim explicó que aún quedan algunos pasos hasta poder hablar de ensayos clínicos, como saber cuanto CDKL5 hace falta añadir, y en qué células en concreto, y cómo de seguro es todo el proceso antes de poder empezar ensayos. Pero como yo lo veo, viendo todo lo que tienen ya avanzado, creo que estamos hablando de un par de años para que la terapia génica legue a ensayos para CDD, y eso en cuanto a plazos de desarrollar nuevas medicinas es básicamente estar a la vuelta de la esquina.
Hay que recordar que dos empresas, Amicus y Ultragenyx, también están trabajando en desarrollar terapias génicas para CDD (aunque no presentaron su investigación en el Foro). Incluso Ultragenyx se llevó este año el Premio de Excelencia del Foro CDKL5 por su contribución preclínica (trabajo en animales previo a ensayos). Con tantos esfuerzos paralelos en marcha confío en que uno o mas de esos programas lleguen a ensayos clínicos.
Luego Kyle Fink nos enseñó los resultados de un proyecto que están llevando a cabo para reactivar la segunda copia del gen CDKL5 que tenemos las mujeres. Como las células masculinas solo tienen un cromosoma X (son XY), las células femeninas (XX) inactivan una de las dos copias para así no producir el doble de niveles de todas las proteínas que están codificadas por genes del cromosoma X. Así que en mujeres cada una de nuestras células aleatoriamente inactiva el cromosoma X que tenemos de papá o el que tenemos de mamá. Y lo que pasa cuando en uno de esas dos copias hay un gen con una mutación es que la mitad de nuestras células estará bien, porque por suerte inactivaron el cromosoma que llevaba la copia mala del gen, pero la otra mitad tendrá deficiencia en CDKL5 porque inactivaron el cromosoma que lleva la copia buena y solo tienen disponible la copia mutada. Lo que el laboratorio de Kyle busca es desarrollar herramientas basadas en la teconolgía de CRISPR que les permitan localizar el CDKL5 inactivo en el cromosoma X que no se está expresando, y rescatarlo sin tocar ninguno de los otros genes que hay alrededor. Es la leche lo que intentan, y hasta hace poco parecía de ciencia ficción. Pero ya lo están haciendo con éxito en células en cultivo (todavía no en ratones con CDD) y consiguen que las células lean las dos copias del gen CDKL5, con lo que si una de las copias está mutada las neuronas tendrán justo lo que necesitan: una copia funcional del gen en cada célula. El paso siguiente es desarrollar la forma de poder poner en el cerebro esos reactivos tipo CRISPR que están usando (posiblemente metiéndolos en virus, como en la terapia génica) y probarlos en ratones.
Y por último David Liu presentó la última frontera de la edición genética: el prime editing. Edición genética es cuando puedes corregir la mutación en el gen. No es poderle a la célula un gen nuevo que le lleva en un virus. No es activar la copia del gen que tenia la célula escondida en el segundo cromosoma X. No. Es llegar a la mutación y arreglarla.
Esta estrategia es la que recientemente publicaron en la revista Nature, y que salió en las noticias de todo el mundo. Lo que el laboratorio de David consigue hacer con su última versión de prime editing es lo que hasta ahora parecía imposible arreglar: los casos en los que al niño le faltan una o dos letras del gen, o se le han insertado una o dos letras en el gen (mutaciones de pérdida de pauta de lectura). Porque ya sabíamos que CRISPR te puede cambiar una letra por otra, darte el cambiazo entre la letra mala y la buena, pero no podía poner o quitar letras. Y ahora David Liu si que puede, y nos enseñó como están empezando a aplicar esta tecnología a células en cultivo con mutaciones de este tipo de CDKL5. Como os decía con el proyecto de reactivación del cromosoma X, lo siguiente sería buscar cómo hacer llegar hasta el cerebro esos reactivos tipo CRISPR que están usando (posiblemente metiéndolos en virus, como en la terapia génica) y probarlos en ratones.
Como estos dos últimos proyectos están en estadíos mas tempranos, yo les calculo más años para llegar a ensayos clínicos que la terapia génica “clásica” de usar un virus con el gen CDKL5. Pero saber que es factible hacer estos cambios en las células, y saber que lo están aplicando ya a CDD, es alucinante y una muy buena noticia para todos nosotros.

Y luego habló el hombre que busca tumbar todos estos plazos, y que nos invitó a todos en el Foro a pensar y trabajar de forma diferente. Se trata de Tim Yu, de Harvard y el hospital infantil de Boston. En el mundo de las enfermedades raras casi todos hemos oído hablar de Mila, la niña con la enfermedad de Batten para que desarrollaron una terapia personalizada en tiempo record para intentar frenar la progresión de su terrible enfermedad. La terapia no es como un fármaco tradicional, sino que se parece mas a un cachito de ADN (oligonucleótido se llama), y lo llamaron Milasen por Mila. Y ese científico que fue capaz de desarrollar Milasen para poder tratar a Mila en tan solo 12 meses desde su diagnóstico es Tim Yu. Al igual que David Liu con prime editing, el desarrollo de Milasen también salió en todas las noticias recientemente. Aprendimos de Tim que no todas las mutaciones son candidatas a este tipo de terapia personalizada con un cacho de ADN, y son sobre todo las que causan defectos de “splicing” en el gen que son candidatas (no muchas de las missense, nonsense o de cambio de pauta de lectura, o cuando falta un cacho del gen). Pero para los casos en los que la mutación es candidata, el laboratorio de Yu está buscando tumbar los plazos tradicionales de desarrollo de medicamentos y poder no ya curar enfermedades de una en una, sino personas con medicina personalizada de una en una (ya que la terapia solo sirve para uno). Esto es una revolución para la medicina, y tenemos suerte de vivir en este momento de la historia en la que estos avances están cambiando la forma en la que practicamos la medicina.

Por cierto que se que muchas familias os liais con saber qué tipo de terapia servirá para todos o qué tipo de terapia es solo para cierto grupo de pacientes. Os he preparado un cuadro de texto con aclaraciones para CDD.
En breve: hemos pasado de decir “cómo podemos hacer que las empresas que trabajan en epilepsia se interesen por CDD” a decir “como podemos tener reemplazo enzimático, Y TAMBIEN terapia génica, Y TAMBIEN reactivación del gen en el cromosoma X, Y TAMBIEN edición genética” para CDD. Y todo en menos de 5 años. Es increíble.
5. DE PRECLÍNICO A CLÍNICO
Una consecuencia de la explosión en la investigación en torno a CDD es que los proyectos han ido madurando y la cartera de programas que se acercan a ensayos clínicos (o que ya están en ensayos) crece a gran velocidad. Esto implica que en las fundaciones y grupos de pacientes nos hemos tenido que poner las pilas y prepararnos para los ensayos.
En la Fundación Loulou hemos estados muy liados este año solicitando la inclusión de CDD en las clasificaciones médicas, apoyando a las empresas que están hablando con agencias reguladoras del medicamento para que tengan toda la información que necesitan, preparando un modelo conceptual de la enfermedad (una cosa un poco técnico de explicar pero que es importante para los ensayos), haciendo estudios sobre medidas que se puedan usar en ensayos clínicos, hablando con empresas farmacéuticas de la posibilidad de que trabajen juntas en solventar los desafíos de los ensayos clínicos en vez de cada uno por su parte, en hacer reuniones con familias para entender mejor lo que valorarían de nuevos medicamentos, e incluso hicimos una reunión con la agencia americana del medicamento (ver mas abajo).
¡No es que hayamos dejado de trabajar en la biología y la investigación en animales! Lo que hemos hecho ha sido expandir el rango de disciplinas en las que trabajamos. En el Foro CDKL5 hicimos 8 reuniones paralelas para tener grupos pequeños de científicos, gente de industria, médicos y pacientes, trabajar en cada uno de los 8 eslabones en la cadena de desarrollos de fármacos, cada uno centrado en un eslabón desde entender que le pasa a las células sin CDKL5 hasta terminar ensayos y pedir la aprobación de un fármaco. Hemos avanzado mucho en cada uno de estos eslabones pero queda trabajo, y para eso estas 8 reuniones paralelas, para identificar lo que falta y los esfuerzos que requerirá.
El que ahora trabajemos mucho mas en las etapas clínicas de desarrollo de fármacos es reflejo de lo mucho que ha avanzado y madurado el campo. Hemos crecido mucho, en tiempo record.
6. LA VOZ DEL PACIENTE, ALTA Y CLARA
Por último, ya que hablamos de haber crecido mucho, debo comentar en el crecimiento de la comunidad de pacientes en CDD. Hemos pasado de arrancar con unas pocas familias, sobre todo en EEUU, Inglaterra e Italia, a tener ahora la Alianza Internacional CDKL5, y organizaciones de pacientes en 18 países incluidos Brasil, China y Japón.
La Alianza organizó su congreso anual en junio, organizado por CDKL5 Inglaterra, y os recomiendo leer sobre este congreso. Conozco muchas comunidades de pacientes y esta es excepcional por lo bien que funciona y lo unidos que están los grupos.
Durante el Foro CDKL5 de este año, miembros de la Alianza actuaron como co-moderadores de esas 8 reuniones paralelas, junto con un moderador de la industria o médicos. Y esto no es algo que se vea mucho, un congreso en el que la comunidad de pacientes esté tan integrada en todas las conversaciones a todos los niveles de la cadena de desarrollo de fármacos, de células a ratones a biomarcadores a ensayos. No tengo duda de que la fuerza de esta comunidad de pacientes es una de las armas secretas del campo de CDKL5.

Y esa fuerza quedó particularmente evidente el 1 de noviembre cuando la comunidad de pacientes de estados unidos se reunió con la agencia americana del medicamento (la FDA) en un Patient-Focused Drug Discovery meeting organizado entre el grupo de pacientes americano y la Loulou Foundation. Un Patient-Focused Drug Discovery meeting (PFDD), o reunión de desarrollo de fármacos centrada en el paciente, son reuniones en las que la comunidad de pacientes se reúne con la FDA para hablar de la enfermedad, y en la que son las familias las que hablan y los reguladores los que escuchan. Este fue sin duda uno de los grandes logros de este año, y podeis ver el video donde los 10 panelistas (padres y abuelos de niñ@s con CDD) y los participantes en la sala explican cómo es vivir día a día con CDD, en que medida impacta a toda la familia, y lo que esperan de futuros tratamientos.
Ya dedicaré una entrada separada al PFDD. Entre tanto quiero destacar que teniendo al menos 7 mil enfermedades raras en el mundo, mas todas las no raras, somos tan solo la enfermedad numero 32 que ha conseguido celebrar este tipo de reunión con la FDA. Y esto refleja la velocidad de desarrollo de terapias para CDD, que ha forzado el interés por la reunión, y la fuerza de esta comunidad de pacientes.
Cuando nació la Fundación Loulou, los padres de Loulou se fijaron como una meta llegar a tener tratamientos (al menos en ensayos) para 2020, y curas para el 2025. Ahora ya sabemos que en 2020 Marinus terminará el ensayo de fase 3 con ganaxolona, y que habremos tenido como mínimo 4 ensayos clínicos en CDD, y colectivamente tratado a más de 150 personas con CDD que podrán beneficiarse ya de estos tratamientos. Y personalmente creo que para 2025 tendremos varias terapias curativas (como la terapia génica) en ensayos clínicos avanzados, y quizás alguna aprobada.
SI puedes imaginarlo, puedes conseguirlo.
Espero que os haya gustado el resumen. Ya me diréis lo que os parece en los comentarios. Y os dejo también el enlace al resumen del Foro de 2018 (en inglés).
Ana Mingorance PhD
Nota: este texto captura mis impresiones de las presentaciones del Foro que más me interesaron como científico y como defensora de los pacientes, no es un texto oficial del congreso emitido por la Fundación Loulou. Escribo estos resúmenes para los padres de personas con CDD, así que a veces me tomo ciertas licencias a la hora de explicar las partes mas técnicas ;-)

MAIN LESSONS FROM THE 2019 CDKL5 FORUM
For the past five years the Loulou Foundation hosts an annual meeting where scientists and drug developers working on CDKL5 deficiency, together with representatives from patient organizations, meet to discuss the latest advances. This was the third Forum I attended, and my second since joining the Loulou Foundation.
Here are the main news and take-home messages from the 2019 CDKL5 Forum that took place in Boston in November 4 and 5.
Versión en Español en este enlace.
The 5th edition of the CDKL5 Forum recently took place in Boston, in November 4 and 5. The Forum is an annual meeting hosted by the Loulou Foundation where scientists and drug developers working on CDKL5 deficiency, together with representatives from patient organizations, meet to discuss the latest advances. This was the third Forum I attended, and the second since joining the Loulou Foundation.

Because of the 5th anniversary, the CDKL5 Forum Director (and my dear friend) Dan Lavery offered an update of how much the entire CDKL5 deficiency field has changed since the first Forum edition. And it has changed so much that it seems we are looking at two different diseases!
So I will borrow Dan’s review of then versus now to share with you an update about the recent CDKL5 Forum and highlight how far along we have come in the CDKL5 Deficiency Disorder field.
1. FROM ULTRA-RARE DISEASE, TO BEING ONE OF THE MOST COMMON GENETIC CAUSES OF EPILEPSY
When the first Forum took place in 2015, CDKL5 Deficiency Disorder (CDD) was thought to affect maybe around 200 children worldwide. It would soon become clear that there were many more cases, but having a specific number has remained a challenge.
This year we obtained the first well-documented incidence estimate for CDD, with mutations in CDKL5 being found in 1 out of every 42,400 live births. The study followed a cohort of all children born in Scotland over 3 years (over 150,000 births), and genotyped all those having epilepsy during the first 3 years of their life. This methodology makes the study very solid, and indicates that CDKL5 would be one of the most common genetic causes of epilepsy in children. Now we know that the real numbers are not 200 cases, but well over 20,000 cases. Definitely not an ultra-rare disease!
The real number of patients diagnosed (prevalence) is still unclear. This is because of two reasons. One, because genetic testing is only regularly done in the recent years, so most adult patients remain undiagnosed even though they exist out there. And two, because we don’t have a good way to track the number of patient diagnosed.
To address this, the Loulou Foundation and IFCR applied this year for CDD to have a unique ICD-10 code, which will allow clinicians to use this diagnostic code with their patients and support the epidemiological studies that are needed. In the meantime, one thing is clear: we are looking for tens of thousands of patients out there, not hundreds.
2. FROM ORPHAN KINASE TO MASTER REGULATOR
If there is one area where the field has evolved dramatically since 2015 is the understanding of what CDKL5 does in the brain. It was clear that it was a kinase, those proteins that turn other proteins on and off as if they had a light switch, but the specific proteins that get turn on or off by CDKL5, and what these do in the neurons, was unknown.

Now scientists have identified many of the target proteins of CDKL5, and produced lab tools (antibodies) that allow us to see when and where CDKL5 is active in the brain. Scientists have also identified that CDKL5 controls a number of proteins associated with the neuronal skeleton (the cytoskeleton) and it is likely through this process that it regulates the presence at the membrane of different proteins including receptors. As a result of this, in the absence of CDKL5 , the formation of synapses (neuronal connections) will remain in more immature stages and the presence of receptors will also resemble the most immature brain state, leading to neuronal hyperexcitability.
There are two important implications for therapies in these results. The first one is that with CDKL5 controlling so many processes it will be hard to “bypass it” or achieve a full compensatory benefit with treatments that target other pathways. We really should try to replace the protein or the gene to achieve the full recovery. The good news is that these treatments are all in development.
And the second important implication is that we have not found large changes in neuronal wiring, or in brain anatomy, and we have certainly not seen neuronal loss or any sign of neurodegeneration. So it would appear that CDKL5 is constantly needed for the very dynamic process of synaptic plasticity and formation. This means that bringing CDKL5 expression back is likely to provide a benefit even in the more mature brains, while in diseases that affect neuronal migration, or that lead to neuronal death, you only have a narrow time window to replace the protein and see any improvement. Based on the biology that we know, I believe CDD will be a good disease for gene therapy or enzyme replacement therapy even in adult patients.
3. FROM ZERO TO FOUR CLINICAL TRIALS AND A GROWING PIPELINE
Another area that has changed dramatically since 2015 is the corporate interest in CDD for the development of treatments. The company who first took the lead was Marinus, and they announced during the Forum that they are on track to soon close their target recruitment for the ongoing Phase 3 trial in CDD, which is 100 patients.
Think about it, we have gone from thinking that there were only 200 patients in the world to being able to run 100-patient trials in parallel to other trials in just a few years.

Because the Marinus trial is not alone, there are three additional clinical trials in CDD going on right now, all in Phase 2 (pilot) stages. PTC recently completed a trial with ataluren for CDD patients with non-sense mutations in CDKL5 and are analyzing the data. Ovid and Takeda have a clinical trial ongoing where they are enrolling around 15 patients with CDD for treatment with their drug TAK-935. And a 10-patient investigator-initiated study with fenfluramine, developed by Zogenix, is starting in New York. During the Forum, Zogenix received the CDKL5 Forum Award of Excellence for “Company Making a Difference” in the clinical space for supporting the latest clinical study in CDD.
So we have gone from having a disease that was thought to be ultra-rare, and that was not recognized as a stand-alone disease by regulators because even the medical community confused it with Rett syndrome, to having a clearly independent unique disease with orphan drug designations and having four clinical trials by five pharmaceutical companies (TAK-935 is developed by Ovid and Takeda together).
And these five companies are not alone. Many companies in the room such as Amicus and Ultragenyx are developing treatment for putting back CDKL5 into the brain, and we were able to see several potential additional therapies presented during the Forum:
A collaboration between the Trinity College Dublin and Insubria University showed that a drug that acts on the neuronal skeleton (which does not work well in the absence of CDKL5) can correct memory problems in mice with CDD.
A group from the University of Pennsylvania showed that another drug, which acts only in immature neuronal receptors that happen to remain present for too long in brains with CDD, also corrects some of the neurological problems in mice with CDD.
The same group also showed that cannabidiol addresses some of the neurological problems in mice with CDD, supporting the data by GW Pharma that Epidiolex could have efficacy in this disease as well.
And even the company Takeda showed a second drug that they are developing (not the one already in CDD trials) and that can also correct some of the neurological problems in mice with CDD by acting on another signaling pathway. They even obtained the Orphan Drug Designation by the FDA for their drug earlier this year.
In total these were four mouse trials showing that there are many more therapies that could move into clinical trials and help reduce not only epilepsy but also other neurological problems associated with CDD. There were some extra treatments presented, but I will cover them in the next section.
What is important to know is that all of these promising findings have been possible because of so much new understanding that we have of how CDKL5 functions in the cell in normal situations, what happens when it is missing, and how CDD presents in mice when their CDKL5 is removed – since it is a bit different than in people. All the investments in research by several patient groups and the Loulou Foundation have made this work possible, and more recently the labs have started being successful at obtaining R01 funding from the NIH. This is one of the areas where the patient community can make a big difference, by supporting the early research and the generation of animal models for the disease that will later allow those groups to be self-sufficient at obtaining the highly-competitive public grants. De-risk early so that a research field can then take off on its own.
4. FROM SYMPTOMS TO CURES
All of the treatments that I listed above in mouse experiments, as well as the current drugs in clinical trials (except for ataluren), help the brain but do not correct the mutation in the CDKL5 gene or the lack of CDKL5 protein in the brain. They help the brain function better with CDKL5.

I have a slide that I use at patient conferences to explain the different types of therapies that could be developed for CDD. That slide is perfect to explain this. Essentially, we know that in CDD the brain does not function well, because it does not have CDKL5 protein, because the CDKL5 gene is mutated. And knowing this we can think of different levels of treatment. We can think of treatments of help the brain function better despite not having CDKL5. Or we could put back the protein. Or we could add a copy of the gene. Or we could go into more difficult approaches and try to correct the mutated copy, or perhaps reactivate the second copy of CDKL5 gene that all females have in their second X chromosome but that is not being used.
In the previous sections I described all the progresses to develop better therapies able to make the brain function better despite not having CDKL5, but where we have seen an explosion of science is in the next approaches: correcting the cause of the disease.

There was a session during the Forum when we had a dream team on stage: Kyle Fink from UC Davis, James Wilson from the University of Philadelphia, and David Liu from the Broad Institute. One by one, they showed us three ways to try to get neurons to produce functional CDKL5, and it was like seeing a conference from the future, when we will be able to correct genetic mutations in a way that we are just beginning to see.
First Jim Wilson presented data from a gene therapy program that he is running at Penn where he has developed a virus that contains the entire CDKL5 gene instead of the virus DNA. When he then injects these virus into the brain of mice with CDD, he can see that the CDKL5 protein is being produced and that this gene therapy corrects some of the neurological problems in mice with CDD. Jim explained that there are still some steps to do before starting clinical trials, like knowing how much CDKL5 is needed, and in which cells, and how safe the entire approach is before it can move into the clinic. But as I see it looks like we are just talking about a couple of years for CDKL5 gene therapy to be ready for trials, and this for medicine is essentially just around the corner.
It is important to know that two companies, Amicus and Ultragenix, are also working very hard for developing gene therapies for CDD (even though they did not present data at the Forum) and that in fact Ultragenyx received the CDKL5 Forum Award of Excellence for “Company Making a Difference” in the preclinical space for their gene therapy program. With so many options ongoing, I trust that one or more of these programs will reach clinical trials.
Then Kyle Fink showed us some data of a project that they are running to rescue the second CDKL5 gene copy that all females have. Because male cells only have one X chromosome (XY), female cells (XX) inactivate one X chromosome so that we don’t produce twice the levels of proteins located in those genes as males do. So all of our cells randomly inactive the X chromosome that we got from dad, and others the one that we got from mum. What happens when one of these chromosomes carries a mutation in the CDKL5 gene is that half of our cells will be fine (they inactivated the “bad” chromosome!) but the other half has deficiency in CDKL5 because they happened to inactivate the “good chromosome” and are left with a mutated CDKL5 gene copy. What the Fink lab is doing is to develop tools based on the famous CRISPR to find the inactive CDKL5 gene in the inactive X chromosome and “release it”, without messing up with any of the other genes in the chromosome. This is very cool, and appeared to be impossible to do until recently. They are doing this right now in cells in culture (not yet in mice with CDD), and the result is that all cells will read both copies of CDKL5, one good and one that is not functioning, and that is perfect because that means that they will all have exactly what they need: one copy that works. Next step is to develop a good technique to get those CRISPR-like tools into the brain (likely into a virus as well) and test it in mice.
Last, David Liu presented what we could call the next frontier of gene editing: prime editing. Gene editing is when you can fix the mutated gene. Not add a new one coming in a virus. Not activating the other copy in the inactive chromosome. But get to the mutated one and fix it.
This approach was recently published in Nature and attractive massive media attention across the globe. What the Liu lab can do with this approach and his latest variations is to correct mutations that until now appeared impossible to correct: those where your child has one or two extra letters in their DNA, or is missing one or two letters in the gene (these are called frame-shift mutations). While we knew that CRISPR would replace one letter, swap the mutated letter by the correct one, we could not insert or delete. Now David Liu can do that, and he told us they are starting to apply it to some mutations in CDKL5. Just like with the Fink approach, the next step after being able to fix the gene in cells in culture is to develop a good technique to get the prime editing tools into the brain (likely into a virus as well) and test it in mice.
Because they are more immature, these last two approaches are likely to take several more years to get to the point when they can be tried in clinical trials. But to know that it is biologically feasible to do these things, and that they are applying it to CDD, is already amazing and tremendously encouraging.

And then there is a man who has been challenging all these timelines, and he too gave a presentation at the Forum and invited us to think about this differently. And that was Tim Yu, from Boston Children’s Hospital and Harvard Medical School. In the rare disease space we all know of Mila, the girl with Batten disease that got a personalized drug developed for her in a record time to try to stop the progression of her terrible disease. The drug is not a traditional type of drug, it looks more like a little piece of DNA (called and oligonucleotide), and got the name of Milasen. And the scientist who developed it and was able to treat Mila just 12 months after her diagnosis was Tim Yu. Just like David Liu with prime editing, this has also been all over the news. We learnt from Tim that not all types of mutations can be targeted with these personalized pieces of DNA, it is mainly the ones that cause “splicing” defects in the gene (not most missense, not the nonsense, not the frameshift, not the ones where a piece of the gene is missing). But for those cases where the mutation would be a candidate, his lab is now pushing the traditional steps of developing a drug for a disease at a time and seeing how to develop the drug for one child at a time. This is a complete revolution, and we are lucky to be living this time where all of these discoveries are changing the way we do medicine.

By the way, I know that many patients and parents get lost with so many genetic approaches and which one would help all patients versus which one is patient-specific, so I have made a separate text box to address this question for CDD.
In short: we have gone from saying “how could get one of the many epilepsy companies to try their drugs in CDD” to saying “how can we have enzyme replacement AND gene therapy AND X-chromosome reactivation AND gene editing all developed for CDD”. All in under 5 years.
Pretty unbelievable.
5. FROM PRECLINICAL TO CLINICAL
A clear consequence of the explosion in research around CDD and the growth of the treatment pipeline is that we have gone from focusing on funding preclinical work (understanding the biology of CDKL5, developing animal models) to having to quickly start working hard on getting the clinical work ready. This means getting ready for clinical trials.
At the Loulou Foundation we have been busy requesting the disease registration in different medical classifications, supporting companies with information as they go through the regulatory process, generating a disease concept model (something that is needed for trials), running studies on clinical outcome assessments (those are the things you measure in clinical trials), discussing with pharmaceutical companies the possibility of working all together to solve the clinical trial challenges as opposed to competing, running meetings with the patient community to understand what they value the most in treatments, and we even hosted a meeting with the FDA together with IFCR (see the next section).
We have not stopped working on the biology and preclinical space! Instead we have expanded what we work on. At the Forum we run 8 separate breakout sessions to have small focused groups of scientists, clinicians and patients go through 8 blocks of what we call “the translational toolbox”, each one focusing on a single topic that combined allowed us to see if we have all we need to take a drug all the way from understanding what happens in the cell to completing clinical trials. We have achieved much in all of these steps but there is still much work to do, so those breakout sessions allowed us to map the needs and focus our efforts in the year until the next Forum.
The greater focus and number of efforts on the clinical space in CDD are a reflection of the maturity of the field. We grew up very fast, in a record time.
6. THE VOICE OF THE PATIENT IS LOUD AND CLEAR
Last but not least, if we are to talk about growth, I must commend on the growth of the CDD patient community. We have gone from having few initial families, organized in the US, UK and Italy, to now have a CDKL5 International Alliance, around 18 national patient organizations, and a world-wide spread that includes countries like Brazil, South Asia, Japan and most recently, China.
The Alliance held a meeting in June, hosted by CDKL5 UK, that I wrote about here and I strongly recommend you reading it if you hadn’t. I have seen many patient communities and this one is exceptional in how well it runs and works together.
During the 2019 CDKL5 Forum, members of the Alliance co-chaired each of the 8 parallel breakout sessions working hand in hand with a clinician, industry professional or scientist chair. This is not something I had ever seen in any other research field, a congress where the patient community is integrated into all discussions along the translational chain, from cells to animals to biomarkers to trials to partnerships. I have no doubt that the strength of this patient community is one of the secret weapons of the CDKL5 field.

And that strength was particularly clear in November 1st, when the US patient community met with the FDA and held an externally-led Patient-Focused Drug Discovery (PFDD) meeting co-hosted between the US patient group (IFCR) and us at the Loulou Foundation. These are meetings where the patient community meets with the FDA to discuss one disease, and where patients speak and the regulators listen. This was a major milestone for the entire field, and I encourage you to watch the video and see the amazing 10 patient (and grandparent!) panelists and the discussions in the room, as the caregiver community explains the regulators how it is to live with CDD, how it impacts the entire family, and the treatment of treatments that they would value the most.
I will dedicate an entire new entry to the PFDD. I just want to highlight here that from all diseases known to mankind, and there are over 7,000 rare diseases alone plus all the non-rare ones, we were the #32 disease-focused PFDD meeting that the FDA had. And that is a reflection of both the fast-growing efforts of the industry to develop treatments for CDD, and the strength of the CDD patient community.
When the Loulou Foundation was born, Loulou’s parents set the goal of treatments (to reach trials) by 2020 and cures by 2025. Now we know that by end of 2020 Marinus will have finished their trial, we will have had at least 4 trials and collectively they will have treated at least 150 kids with CDD that will be able to benefit from these treatments. And I personally believe that by 2025, we will have multiple cures (gene therapies and similar) in advanced clinical trials, possibly even some approved.
If you can imagine it, you can achieve it.
I hope you enjoyed this summary! let me know your thoughts in the comments. Here is my article on the 2018 Forum meeting.
Ana Mingorance, PhD
Disclaimer: These are my own impressions from the presentations that I was most interested in as a scientist and patient advocate, and not an official text about the Forum by the Loulou Foundation. I write these texts with the parents of individuals with CDD in mind, so excuse also my lack of technical accuracy in parts ;-)

Dravet syndrome drug development pipeline review 2019
The 2019 Dravet Syndrome Pipeline and Opportunities Review provides a review and analysis of 12 drug candidates in development for the treatment of Dravet syndrome, including 11 products that have received orphan drug designations. The Report includes the most recent updates on programs from GW Pharmaceuticals (Epidiolex / Epidyolex), Zogenix (Fintepla, ZX008), Biocodex (stiripentol) , Ovid Therapeutics (Soticlestat, OV935, TAK-935), Takeda Pharmaceutical, Supernus Pharmaceuticals (SPN-817, Huperzine), Xeris Pharmaceutical (diazepam), Epygenix Therapeutics (EPK-100, -200 and -300), NeuroCycle Therapeutics (NCT10015), PTC Therapeutics (ataluren), Stoke Therapeutics (STK-001), Encoded Therapeutics and OPKO Health (OPK88001, CUR-1915).
It has been a year since we released the 2018 Dravet Syndrome Pipeline and Opportunities Review, a market research publication that provides an overview of the global therapeutic landscape of Dravet syndrome.
In the last 12 months the pipeline has changed by, among others, the approval by the FDA of Diacomit (stiripentol), the CHMP positive opinion on Epidyolex (cannabidiol) approval, the IPO from Stoke Therapeutics and the arrival of a new company pursuing a gene therapy approach for Dravet syndrome (Encoded Therapeutics).
The 2019 Dravet Syndrome Pipeline and Opportunities Review provides a review and analysis of 12 drug candidates in development for the treatment of Dravet syndrome, including 11 products that have received orphan drug designations.
The Report includes the most recent updates on programs from GW Pharmaceuticals (Epidiolex / Epidyolex), Zogenix (Fintepla, ZX008), Biocodex (stiripentol) , Ovid Therapeutics (Soticlestat, OV935, TAK-935), Takeda Pharmaceutical, Supernus Pharmaceuticals (SPN-817, Huperzine), Xeris Pharmaceutical (diazepam), Epygenix Therapeutics (EPK-100, -200 and -300), NeuroCycle Therapeutics (NCT10015), PTC Therapeutics (ataluren), Stoke Therapeutics (STK-001), Encoded Therapeutics and OPKO Health (OPK88001, CUR-1915).
The 2019 Dravet Syndrome Pipeline and Opportunities Review also includes an analysis of the competitive landscape, unmet needs, and evaluates current and future opportunities of the Dravet syndrome market.
The report is now available in this site.

What the CDKL5 Deficiency community can teach us about patient centricity
I used to say that at the patient communities “we set the agenda”. It turns out we didn’t, we were borrowing the agenda from scientific meetings. The 2019 CDKL5 Alliance International Research and Family Conference redefined what a patient-centered conference truly is. In this article I summarise the elements that make a meeting truly patient-centered.
The first time that I attended a meeting where scientists and patients shared the room was about 12 years ago. It was a small conference on spinal cord injury, and I was an academic postdoc. I remember a man with spinal cord injury, who took the stage and addressed the scientists in the audience. He said “some of you see the glass as half full, and some of you see it half empty. I am here today to ask you that regardless of where you stand on the argument, just grab that glass and do something about it”.
That was my first exposure to an impatient patient.
Some years later I was a scientist at a pharmaceutical company, and we were asked to present an update on our projects to a panel that included scientists, physicians, investors and also some patients. As they all introduced themselves going around the table with the usual line “I’m Dr X and have a PhD in Pharmacology and blah blah”, they reached the man with Parkinson’s disease. He said “I’m Mr X and 6 years ago I was diagnosed with Parkinson’s disease. I feel I’ve earned multiple PhDs on Parkinson’s by now”. He was very right, he was the most knowledge person in that panel.
That was my first exposure to a patient expert, who knew more about a disease than the biotech investors and the physicians sitting next to him.
Last weekend I attended a Research and Family meeting organized by a rare disease patient organization. As a rare disease scientist and patient advocate, I have attended many of these meetings, yet this was my first exposure to a real patient-centric conference. Anything else I saw before, in those 12 years, was a conference that included patients, often organized by patients, but a complete different beast. Let me explain what I mean by this as I review the 2019 CDKL5 Alliance International Research and Family Conference.

2019 CDKL5 Alliance International Research and Family Conference
In June 22nd and 23rd 2019, the CDKL5 Deficiency Disorder (CDD) patient community got together in Edinburgh, UK, for a joint conference with families, clinicians, scientists and pharmaceutical companies.
This one is, for me, a very special patient community. A decade ago, most families with a child diagnosed with CDD were told it was an ultra-rare disease and that they were the only one in their country, or one of a handful at most. Today we know that the disease affects tens of thousands of people, and there is an international patient Alliance that includes 17 national patient organizations and a few other parent-led groups. I’ve been part of this community for the past year and a half since I joined the Loulou Foundation.
There were families from most countries in the Alliance present at the Conference, including many bringing their children with them. Scientists, clinicians and company representatives were outnumbered by the families.
It is only while attending this meeting that I realized that all other patient-organized conferences that I had attended followed one of two formats:
Internal (one country) patient organization meetings, where the focus is the organization itself (accounts, the projects that they finance, priorities)
Broader meetings where clinicians offer updates on clinical trials and scientists offer updates on research to an audience of patient families.
This latter type is the most common. Clinicians and scientists often, but not always, dumb down their explanations so that the families can follow, but other than that it is exactly the same thing that you would expect from a scientific meeting. In many cases the agenda is shaped by the medical or scientific advisors from the patient groups. In other cases, it is designed by the patient organization, but it is still very much inspired in the way scientific meetings are run.

Carol-Anne Partridge, CDKL5 UK
But not the latest CDKL5 Alliance meeting…
The program of this conference was designed by two exceptional people. One is Carol-Anne Partridge, Chair of the host organization, CDKL5 UK, and new Chair of the International CDKL5 Alliance. The outgoing Chair of the Alliance, Rick Upp, introduced Carol-Anne as “one of the 5 Fundamental Forces of Nature” and he is very right. Her stubbornness and determination mean that a meeting organized by CDKL5 UK could not be a conventional meeting.
And Carol-Anne met her perfect match in Philipp von Gallwitz, also a rare disease dad. Philipp is a former pharmaceutical industry professional who now specializes in bridging the patient world and the pharma world. Together they made a program where the focus is on the families attending the meeting, not on the science and clinical presentations.
Once of the scientists in the room described the conference as “celebrating a birthday, or the New Year”. That is how special it was.
Elements of a patient-centric conference
1. The meeting should have a clear mission which is not to update families on what scientists and physicians are doing
The first page of the conference agenda made it clear that the meeting had a specific purpose, and not just to get updates for the families. It read:
“ The main objective is to support families, physicians and industry get ready for clinical trials. Unless otherwise indicated, all information and discussions shall be such that families can understand and contribute. All sessions shall clarify why the presented content is important for families and what they can do in practice to support. Conference moderation will strive to finish all sessions with specific actions and next steps”
The purpose of the conference was to work together towards clinical trial readiness, with patients as active members, not passive listeners. We had to come out of that weekend with a list of clear actions and make the most out of the time we were all together. And so we did.
2. The meeting should start and conclude with families on stage
Right after the host welcomed everyone to the meeting, a mum of a child with CDD explained their story. It was a difficult story for everybody, since she had lost her little boy. It also reminded all why we were in the room: she explained how she shared other families’ pain as if it was her own, and also shared all of their happy moments. That helped us all remember that we are a community and why we were there.
The last speaker of the conference was a grandfather. He was also a great speaker, and helped us see the impact of diseases even beyond the parents and siblings. As in a good speech, opening and concluding the meeting with a common element helped bookend the conference and close the circle around patients.
3. Prepare your presenters to communicate what is important
Philipp made sure that all presenters were briefed about the mission of the meeting and to coach them and review their presentations prior to the conference. Scientists are often used to giving talks where they want to impress other scientists with their hard work and knowledge. Philipp made sure they knew that despite having more scientists in the room, the target audience were the families and the presentations had to clearly explain the medical problem that they were researching, what they found, what it means to the patients and how can families or the community help them. And to do all this in simple terms.
Asking them to simplify the language was key for the meeting success. For example you might love the word “microtubules”, but it works a lot better if you can talk about “the highways along the neurons” as well as the traffic consequences of the mutation.
I sometimes find that families are better at explaining medicine than we (scientists) are. A mum explained to me Cortical Visual Impairment (or cortical blindness), a visual problem in CDD kids due to neuronal problems, not eye problems, as “it is as if the TV works perfectly and the problem is that it is plugged into a non-functioning outlet”. I will remember that one.
4. Use the meeting to build the future: every presentation, panel or discussion should focus on how the patient community can advance the field together with the other stakeholders
Often patient conferences are dominated by scientists and clinicians, with patient families becoming the listener and observer as the professionals explain them what they have been doing “for the patients”. This conference was designed to be different.
Every speaker was instructed to explain to the families what they might need from them: clinical data, blood samples, funding, feedback on their medical scale….
We had multiple sessions where we discussed how the patient community can contribute to advancing the field and how to work together. For example: how can we ensure that even basic research is relevant to patients, how can the patient community provide sufficient support to researchers and physicians, how can we involve families better in trial design and recruitment, etc
Every session was targeted towards identifying practical relevant priorities, actions, quick wins, strategies…. That focus on actionable elements means we all went home with an entire notebook of actions and that the CDKL5 Alliance will have a very busy year!
5. Maximize the time and opportunities to listen to the patients / families
At the 2019 CDKL5 Alliance International Research and Family Conference there was no audience. There were only participants.
Everybody in that room had multiple opportunities to contribute, whether they were patient families or scientists or company representatives.

Patient Experience Workshop
We had a panel session where the entire room was the panel. We discussed what researchers need from families and what families need from researchers. We learned wonderful things that I’m sure wouldn’t have come up in a more structured meeting. For example I learned that because there are only 6 patient families in Ireland, they felt they were not enough to come together and organize an annual meeting, but ever since there was also a research lab in Dublin dedicated to CDKL5 the families felt it was the perfect reason to come together with the scientists and host their first annual meeting. The academic lab helped create the momentum that the small patient community needed.
We had a World Café, where all of the participants divided in 4 groups an discussed the main challenges, opportunities and role for patients and professionals in research, development, clinical care and clinical trials. Every single person participated in a brainstorming on each of the 4 topics, and contributed to a list of action items and a roadmap.
We had a patient experience workshop where all families were invited to provide their testimonies on the symptoms and impacts of living with CDD and their hopes and perspectives on treatments. It was shaped as a mini-PFDD meeting, where we only listen to the caregivers in a facilitated discussion, and it was the most amazing experience I’ve had a patient conference. Many of the families also told me how it had been the highlight of the day, and I will try to incorporate similar sessions in future patient conferences. For the industry professionals present at the meeting it was also an exceptional opportunity to learn how to see the disease through the eyes of the patient families, and how different it can be from the simpler clinical description of the disease in medical publications.
There was truly no audience at this conference, only participants.
6. Incorporate the community leadership

“There are 5 Fundamental Forces of Nature: gravity, weak nuclear force, strong nuclear force, one that I can never remember, and Carol-Anne Partridge” . Rick Upp, about Carol-Anne Partridge
The CDKL5 Alliance also hosted a closed meeting with all of the representatives from the national CDD patient organizations. Every year, the country hosting the international conference becomes the next Chair of the Alliance. So just as the UK brexits away from everybody else, CDKL5 UK takes the leadership of this exceptional patient community from the previous US Chair.
The mission of the Alliance is to collaborate and support each other, identify more patients, set up new clinical trial sites and centers of excellence, and overall to support the medical and research community as we progress towards new treatments for the disease. Running an annual international meeting the size of this one is not easy, but it is an exceptional opportunity to bring all of the organizations together under the same roof and keep it together and productive.
For the CDKL5 Alliance, the new year started on June 23rd, and I look forward to supporting them in these next 12 months.
Final notes
I personally love family conferences. I love to get to meet many new families and learn from them. I loved meeting Eric, who can’t yet speak or hold his head up but seriously likes traditional Scottish music, and to meet families from my home country. I get frustrated in these conferences at the slow pace of drug development, but I also get much more energized and inspired so the net effect is very positive.
I used to say that at the patient communities “we set the agenda”. It turns out we didn’t, we were borrowing the agenda from scientific meetings.
Thank you Carol-Anne and Philipp for truly setting up the agenda, and redefining what a patient-centric conference is truly about.
Let me know your thoughts in the comments,
Ana Mingorance, PhD
Image credits: some are my pictures, some are pictures other participants took and shared in social media

Clinical trials in CDKL5 Deficiency Disorder – 2Q 2019
There are currently 4 clinical trials ongoing or about to start in CDKL5 Deficiency Disorder: ataluren, ganaxolone, TAK-935 and fenfluramine. This article is a summary of where we are with clinical trials for CDKL5 Deficiency Disorder for families and other interested readers including what we know about these four drugs, their efficacy, at which level of clinical development they are at, and where can you learn more about these trials.
Some rare diseases can go for many years without much research on them. Other rare diseases, however, attract so much attention from scientists and companies that in a couple of years can make the progress that would have otherwise taken decades.
This is the case of CDKL5 Deficiency Disorder (CDD), a monogenetic rare disease that affects brain development and causes a very severe epilepsy with hundreds of seizures a month. I also reviewed all of the news on CDD in my review of the last CDKL5 Forum meeting HERE (October 2018).
This article is a summary of where we are with clinical trials for CDKL5 Deficiency Disorder for families and other interested readers.
FIRST, A NOTE ABOUT CLINICAL TRIALS
There are currently 4 clinical trials ongoing or about to start in CDD.
If you are coming from the patient side, and not from the medical community, you have probably heard about clinical trials being always divided in three stages:
Phase 1 trials: Small trial (study) in healthy adult volunteers, not in patients. The purpose of this phase is to determine the safety of the drug, as well as to explore different doses of the drug and measure the biodistribution (how soon you eliminate it, how it breaks down, does it accumulate… etc)
Phase 2 trials: Also known as “pilot” trials. Small trials in real patients with the disease that the drug intents to treat. This phase is mainly intended for determining safety, and also to pick a sign of efficacy. There are usually not enough patients to be sure that the drug works, but it enables companies and regulators to decide to move on to the next phase if the data looks good.
Phase 3 trials: Also known as “pivotal” trials. These are large confirmatory studies, in patients, with a placebo-controlled group (to serve as a control of what the normal change in the disease would have been in the absence of the drug, everything else remaining the same). You often need two of these trials for approval. Depending on the disease, it could be over a thousand patients per group. Other times it is many hundreds. Because of the need of finding so many patients it can take years to complete.
This is the default design, but there are exceptions to it. One exception is that when diseases are rare, you often need fewer patients, and might only need one pivotal trial to show efficacy and get a drug approved. This means a company can go from starting clinical trials to approval in 4 years, as GW Pharma recently did with Epidiolex for Dravet and Lennox-Gastaut syndromes, as opposed to 10+ years on non-orphan diseases!
Another exception is when the drug being tested has already gone through a Phase 1 evaluation in the past when being considered for other diseases, so once the company that owns the drug shows an interest in your disease they can move straight into Phase 2 (pilot) trials.
Both exceptions are true for CDD, so as you will see below our timelines are much faster than the usual length of Phase 1 + Phase 2 + Phase 3 trials that you will find described in most on-line materials. Also, our pilot (Phase 2) trials need less than 20 patients, and the first drug that has reached the pivotal (Phase 3) stage only needs one trial with 70 patients. This makes it all a bit more doable and a lot faster.
One important note about CDD trials is that all of them add the experimental drug or the placebo treatment to the other medications that the patient is already taking for a duration of 12 to 17 weeks. This means that no participant will ever find themselves in a “no treatment” trial group if receiving placebo, they will simply start adding the actual experimental treatment to their usual medications later. Some of the clinical trials do not even include placebo group.
So let’s jump into the update: which are these four drugs, what do we know about their efficacy, and where can you learn more about these trials or what comes after them.
CDKL5 DEFICIENCY DISORDER CLINICAL TRIALS REVIEW

From the drug that first started clinical trials in CDD to the one that is about to start, these are the four clinical trials for CDD that you should know about:
#1 ATALUREN – PTC THERAPEUTICS
Ataluren is a drug approved in Europe for the treatment of a subset of patients with Duchenne Muscular Dystrophy and marketed under the name of Translarna. It is currently completing a placebo-controlled Phase 2 (pilot) study at NYU Langone Medical Center in children with CDKL5 Deficiency Disorder caused by non-sense mutations.
How does this drug work?
The brand name of ataluren is “Translarna” because it facilitates translationof RNA. I like how clever the name is. What it does is to target specifically a type of mutations known as non-sense mutations, which cause a premature stop in the gene sequence. These mutations appear in many different genes. That is why we can test this drug in diseases other than Duchenne, such as CDD and Dravet syndrome (the second disease evaluated in the same clinical trial). What ataluren does is to make the cell read through that premature stop and complete the protein that is otherwise missing in that disease.
Is there any previous clinical experience with this drug?
Yes! Ataluren is already approved for Duchenne, so the active dose and the safety profile are known. There is still no efficacy data in patients with neurodevelopmental diseases or epilepsy because these CDD and Dravet syndrome trials are still ongoing. One important difference with Duchenne is that CDD and Dravet are diseases of the brain, not the muscles, so it is possible that the drug doesn’t get well-enough into the neurons to work in these neurodevelopmental diseases. We will have to wait for the trial results to know that.
How is the clinical trial in CDD?
The trial is a pilot study, involving 9 patients with CDD that all go through some months in treatment and some months in placebo. This is called a “crossover” design, where some patients start in the drug and are then crossed over to placebo, and others start in placebo and are then crossed over to drug. This way, none of the patients in the study has to be only in placebo, and it also means that each patient is their own control.
The trial measures epilepsy as the main symptom for improvement. They also track cognitive function and quality of life as additional potential areas of improvement.
What would be the next steps for approval?
This trial is only taking place at one hospital, NYU Langone Medical Center, because it is what is known as an “investigator-initiated study”. Investigator-initiated means that the company that owns ataluren was not who decided to start this trial, but the investigator (the clinician) from NYU approached the company and asked them to let them run this clinical trial at their hospital.
Because it is a small pilot trial, the resulting data will not be sufficient for requesting approval of ataluren for treating CDD. So first the drug results need to be published, it has now completed recruitment but the data collection and publication are not yet completed. And after that, if the data is positive, PTC would need to run a Phase 3 pivotal trial. So the next step to this clinical trial is another (larger) clinical trial if the results are positive.
Where can I find more information about the trial?
The clinical trial and contact information are here.
#2 GANAXOLONE – MARINUS PHARMACEUTICALS
Ganaxolone is a drug currently in Phase 3 (pivotal) trials in CDD. It is also in clinical trials for other neurological diseases. It has not yet been approved for any disease, and if everything goes well it is likely to become the first drug to be ever approved for the treatment of CDD since it is the most advanced one.
How does this drug work?
You might be familiar with medications like Valium and Xanax. They belong to a class of drugs, benzodiazepines, that are used for anxiety, insomnia and muscle relaxation among other uses. Some drugs in this class, like clobazam (Onfi) are even used for treating epilepsy. These drugs all enhance the activity of a type of brain receptors called GABA receptors and the result is “brain relaxation”. The brain of people with CDD has an excess of neuronal activity, so drugs that enhance or facilitate GABA receptor function can help reduce excessive activity and minimize some of the symptoms.
Ganaxolone binds to the GABA receptor and enhances their activity in a different way to how benzodiazepines work, so it is thought to achieve the “brain relaxation” with slightly different properties. It is also known that in some epilepsy syndromes, patients have low blood levels of a ganaxolone-like endogenous body hormone, so part of the efficacy of ganaxolone could be due to it helping correct this deficiency.
Is there any previous clinical experience with this drug?
Yes. Ganaxolone has been in clinical trials for other neurological diseases, like Fragile X syndrome and partial onset (focal) seizures in adults. More importantly, there has been a Phase 2 (pilot) trial in people with PCDH19 epilepsy and CDD that sowed it had efficacy on both patient populations when looking at their epilepsy. Because ganaxolone was also safe, it has now been progressed to the final Phase 3 (pivotal) studies in PCDH19 epilepsy and in CDD, now in two separate studies.
How is the clinical trial in CDD?
The Phase 3 clinical trial of ganaxolone in CDD is called the Marigold Study and takes place at numerous centers internationally. The trial is currently recruiting for patients. You can find more information about trial sites in the website of the Marigold study.
To enroll in the trial the patient needs to have a mutation in CDKL5, be 2-21 years old, and have at least 16 “major seizures” per month, which includes tonic-clonic seizures and atonic (drop) seizures. They are looking for at least 70 trial participants.
During the trial some of the patients are given ganaxolone while some receive placebo. Neither the families nor the physicians know which group the patient is in. After 17 weeks all patients are offered a chance to take ganaxolone, so if your child is placed in the placebo group it just means they will be starting the actual treatment about 4 months later.
The trial will measure epilepsy as the main symptom for improvement, and will also track improvement in other areas such as attention and behavior.
What would be the next steps for approval?
The Marigold study is a pivotal trial, meaning that it is a final trial. Once the study is completed, Marinus will submit all the documentation to the different regulatory agencies and request the marketing authorization for the treatment of CDD.
Where can I find more information about the trial?
Marinus has created a website specifically for this trial: the Marigold study. You can also find more information as well and some CDD materials for patients at the company website.
#3 – TAK-935 / OV935 – OVID THERAPEUTICS AND TAKEDA PHARMA
TAK-935, also known as OV935 since it is co-developed by Takeda and Ovid, is a drug currently in Phase 2 trials in CDD and other neurodevelopmental syndromes with epilepsy. It is an experimental drug and it is not yet approved for any other disease.
How does this drug work?
As you will remember from some paragraphs before, GABA is an inhibitory substance in the brain, which is why some drugs like ganaxolone enhance the activity of the GABA receptors to reduce brain excessive activity. They enhance brain inhibition. The excitatory substance in the brain is glutamate, and we know that the brains of people with CDD have an excess of neuronal activity in part due to too much glutamate signaling. What TAK-935 does is to reduce this excessive activity by reducing glutamate signaling, therefore bringing brain activity down to healthier levels. It reduces brain excitation.
Is there any previous clinical experience with this drug?
TAK-935 had already been in clinical trials for other neurological diseases although it was never taken all the way to the market. Because of that, the Phase 1 trials were already done. When Ovid and Takeda decided to test TAK-935 in drug-refractory epilepsies they run a Phase 2 (pilot) trial with 18 patients with a variety of rare epilepsy syndromes. Because the safety was good, and the patients experienced a reduction in seizures, the companies decided to progress the drug to further testing, and it is now being studied in four different Phase 2 (pilot) trials: one for Lennox-Gastaut syndrome, one for Dravet syndrome, one for Dup15q syndrome, and one for CDD.
How is the clinical trial in CDD?
The CDD and Dup15q studies are combined under a trial called the ARCADE study. The trial is currently recruiting patients, and is looking for 15 people with CDD ages 2 to 35 with at least 3 motor seizures per month.
Because it is a pilot study, there is no placebo group. All 15 participants will receive the experimental drug on top of their regular baseline medication. Participants will take TAK-935 for 20 weeks (8 weeks bringing up the dose slowly followed by 12 weeks at maintenance levels), and the total trial duration beginning to end is 30 weeks. At the end of this period all participants will be offered to keep taking the drug if they found it to be effective.
The trial will measure epilepsy as the main symptom for improvement, and will also track general improvement.
What would be the next steps for approval?
Because the clinical trial is a Phase 2 (pilot) trial, and not a final pivotal trial, the results of the trial will not be sufficient for the companies to request a marketing authorization. If the trial results are good, then they will have to run one or two Phase 3 (pivotal) clinical trials like the one currently ongoing with ganaxolone, involving many more patients and most likely a placebo group. So the next step to this clinical trial is another (larger) clinical trial if the results are positive.
Where can I find more information about the trial?
You can find more information about this trial in the ARCADE study website.
#4 - FENFLURAMINE – ZOGENIX
Fenfluramine is a drug that was approved many years ago for treating obesity as part of a combination pill. It was later discovered to have efficacy in treating drug-refractory epilepsy in an epilepsy syndrome called Dravet syndrome, and it has now completed all of the clinical trials for Dravet syndrome and is awaiting regulatory review to obtain the marketing authorization. Pilot studies have shown that fenfluramine has efficacy in other syndromes as well, so a pivotal trial is ongoing for Lennox-Gastaut syndrome and the company is interested in evaluating the efficacy of their drug in other syndromes with epilepsy to identify potential new diseases that could benefit from it. One of those, is CDD.
How does this drug work?
While GABA and glutamate are respectively the main inhibitory and excitatory substances in the brain, there are several other substances that are known as “modulatory” because they tweak brain activity in many different ways. One of these is serotonin, and you might be familiar with antidepressants increasing serotonin signaling to stabilize people mood. This is also what fenfluramine does. Fenfluramine enhances serotonin signaling through some of the serotonin receptors, of which they are in total 15 different ones each playing different roles in the brain. It is not clearly known why these serotonin receptors are involved in epilepsy, but the clinical data so far has shown that fenfluramine is a very good anti-epileptic drug, at least in children with Dravet syndrome. Fenfluramine might also bind to other receptors in the brain, this is all still being studied.
Is there any previous clinical experience with this drug?
Yes! The reason why fenfluramine will be studied in CDD is because of the results they have seen with Dravet syndrome. Dravet syndrome is another neurodevelopmental syndrome with epilepsy, and unlike CDD where some of the patients ultimately outgrow their seizures, in Dravet syndrome this doesn’t happen. Two Phase 3 (pivotal) trials with fenfluramine in these children showed that fenfluramine could reduce seizure frequency by more than 70%, and about one in four participants was seizure free or “near seizure free” (about 4 seizures a year). The side effect profile of fenfluramine is similar to other anti-epileptic drugs and it requires extra cardiac monitoring.
How is the clinical trial in CDD?
The trial is a pilot study, involving 10 patients with CDD that all receive the drug because there is no placebo group. The trial has not yet started recruiting, it will take place at NYU Langone Medical Center, and is looking for children with CDD ages 2 to 18 with more than 4 convulsive seizures a month that will receive the drug added to their baseline medication during 14 weeks.
The trial measures epilepsy as the main symptom for improvement. They also track quality of life and general improvement.
What would be the next steps for approval?
This trial is only taking place at one hospital, NYU Langone Medical Center, because it is again an “investigator-initiated study” as explained above for ataluren.
Because it is a small pilot trial, the resulting data will not be sufficient for requesting approval of fenfluramine for treating CDD. So the next step to this clinical trial is another (larger) clinical trial if the results are positive.
Where can I find more information about the trial?
You can find more clinical trial and contact information HERE, and additional information about previous results with fenfluramine in Dravet and Lennox-Gastaut syndromes HERE.
THE FUTURE - IS THERE ANY MORE RESEARCH ONGOING BEYOND THESE TRIALS?
Oh yes! These four are all therapies that were already developed for treating other diseases (most for epilepsy, ataluren for addressing one specific mutation type). This means that they could move really fast into CDD, and start clinical trials right away without needing more research or improvements on them.
But there are other very exciting programs that have been started specifically to address the genetic cause of CDD. These ones will still take a couple of years before they can start clinical trials because they have been started from scratch to be designed for CDD, and that takes time that we didn’t have to wait with the “ready for clinical trials” drugs.
As a reminder of how CDD happens, CDKL5 is both the name of a gene and the protein that it produces. Each protein in the body has a specific function. The CDKL5 protein function is to put a phosphate onto other proteins which is like an on/off switch for those other proteins. This allows CDKL5 to turn on and off many functions of the neurons. Proteins that do this are called enzymes.
We are all born with mutations that were not present in our parents, that is how evolution works, but in most cases these mutations are in non-important regions, or at least are not too damaging. When one of these mutations, however, happens in the CDKL5 gene sequence and either breaks the sequence or gives it the wrong instructions, then that person cannot produce the CDKL5 protein or produces a non-functional version. Without good CDKL5 protein, all of those functions in neurons that needed CDKL5 to put all of the on/off switches in the right configuration are now not functioning properly. This is why the deficiency in CDKL5 is so bad for the brain.
There are two main efforts to fix these problems in patients with CDD. Not to treat their symptoms, but to correct the faulty biology that is causing the symptoms. These are the type of approaches that in the patient community we often call cures, although you will not hear this word from pharmaceutical companies. They prefer to call them disease-modifyingtreatments because they change the disease.
The first approach is gene therapy. If you could give each neuron a new copy of the CDKL5 gene then they will be able to produce the protein and function as a normal cell. There are multiple efforts going on in this area, but I will highlight the program from the company Ultragenyx, who announced last October that they will develop a virus carrying the CDKL5 gene as a gene therapy for CDD.
The second approach is to simply add to the brain the CDKL5 protein. This has been done in the past in other diseases caused by enzyme deficiencies, and are known as Enzyme Replacement Therapies. I would highlight here that the company Amicus has been working on this approach for the last couple of years, trying different approaches to deliver the CDKl5 protein to the brain.
This means that in the next couple of years we will have several clinical trials for CDD that target disease symptoms, and then we will start having clinical trials with therapies that target the cause of the disease. The first group of drugs will reduce the symptoms of the disease and give the patients a chance to acquire more skills faster while they have less seizure burden. The second group of therapies, in particular when used in very young kids, will lead us to a future where children born with mutations in CDKL5 will pretty much be able to grow as if their gene was not mutated. As usual in medicine some trials will fail, but other trials will get started. The important message is that there are so many programs ongoing that the question is not IF we are going to make it to effective therapies, but WHEN.
Let me know if you have some questions on these trials that I didn’t cover in the article!
Ana Mingorance, PhD

Main Lessons from the World Orphan Drug Congress USA 2019
Many orphan drugs are advanced therapies. Pricing and access are major issues. Epilepsy is catching up with gene therapy. We shouldn’t call them rare diseases, but frequently misdiagnosed diseases. Either we wait 2,000 years for treatments or we start thinking “many diseases at a time”, and online patient communities are now part of the drug development process. That’s the short summary of the main lessons I took home from attending the World Orphan Drug Congress at the National Harbor April 10-12. The WODC one of the largest meetings dedicated to the development of new medicines for rare diseases and takes place once in the US and once in Europe every year. In a bit more detail, here is the expanded list of what I would like to share with you from the conference.
Many orphan drugs are advanced therapies. Pricing and access are major issues. Epilepsy is catching up with gene therapy. We shouldn’t call them rare diseases, but frequently misdiagnosed diseases. Either we wait 2,000 years for treatments or we start thinking “many diseases at a time”, and online patient communities are now part of the drug development process.
That’s the short summary of the main lessons I took home from attending the World Orphan Drug Congress at the National Harbor April 10-12. The WODC one of the largest meetings dedicated to the development of new medicines for rare diseases and takes place once in the US and once in Europe every year.
In a bit more detail, here is the expanded list of what I would like to share with you from the conference:
1- A lot of “orphan drugs” use new technologies.
Although the Orphan Drug Act was conceived to stimulate development of therapies for rare diseases in general, and it has often been used in an “opportunistic way” for drugs that could have targeted broader populations, the new technologies that allow us to target specific gene defects are taking over the field. There was an entire day on Next Generation Therapies that highlighted programs in development using antisense, base editing, gene therapy and cell therapy approaches. There was also a great plenary session with multiple of these approaches including companies already in the clinic such as Alnylam, Sangamo Therapeutics and CRISPR Therapeutics.
I believe that with most rare diseases having a genetic cause, and most genetic diseases being rare diseases, it is only natural that gene-targeting technologies will find their home in the rare disease space.

2- Big challenges: delivery, manufacturing and drug pricing
In many of the debates the conversation went beyond the medical use of these new technologies and focused on some important challenges that the orphan drug community needs to face and address. One important issue that multiple of the new technologies face is how to get into their target tissue. This is particular complex for neurological diseases, although some companies are making great progresses in this space and Alnylam announced a few days before the WODC a partnership with Regeneron to apply their RNAi therapeutics to ocular and neurological diseases. Manufacturing challenges also limit cell and gene therapy development, making many of these one-time treatments too expensive. And that was indeed one of the main topics of discussion in the meeting: pricing and affordability of these new therapies. Emil Kakkis from Ultragenyx stressed the importance of ensuring access to therapies saying that this is the golden age for rare diseases and yet a therapy means nothing if patients cannot get access to it. While delivery and manufacturing are technical challenges, identifying suitable payment mechanisms that guarantee patients access to treatments is a social challenge that is likely to be the hardest to address.
3 – Epilepsy meets the future
I regularly participate in orphan drug conferences, where I get to see all these exciting new technologies, yet at the epilepsy conferences (my field) most of what we see is traditional pharmacology that focuses on reducing brain activity without targeting the specific cause of the epilepsy in that patient. I work on neurological syndromes with epilepsy, and a very large number are monogenetic meaning that they would be the ideal target for these new approaches. Barry Ticho from Stoke Therapeutics said it very clearly at the WODC:
“there are more than 100 epilepsy genes but not a single therapy that treats the causes”.
But that is changing.
At the WODC we could see multiple approaches in development to specifically target the causes of several neurodevelopmental syndromes with epilepsy. In addition to Stoke presenting their antisense approach for Dravet syndrome, the small company RogCon presented their antisense approach for SCN2A epilepsies, Roche presented their antisense approach for Angelman syndrome, and as part of a panel, Xenon Pharma highlighted their small molecule approach with a potassium channel opener for KCNQ2 epilepsy. This is a good reminder that for some genetic diseases, small molecules could also offer an excellent disease-targeting approach, as also seen in cystic fibrosis where different CFTR protein alterations are treated by specific molecules designed to address those alterations.

To prepare the field of epilepsy for these new personalized approaches, early genetic diagnostic is paramount. Invitae has now extended the Beyond the Seizure epilepsy program to offer free genetic testing to all US children under 60 months of age with epilepsy. This program is sponsored by BioMarin, Stoke and Xenon, and I hope it will get more sponsorship to be opened to patients currently older than 5 years old and from other countries. Having a genetic diagnosis (early or not) is still one of the big challenges for rare disease patients around the world.
4- Don’t call them rare – the importance of the words we use
Carol-Anne Partridgeis the Founder of a patient charity, and the mother of a beautiful girl with CDKL5 Deficiency Disorder. In her presentations, Carol-Anne always stresses that we need to change the narrative around rare diseases. Her experience resonates with that of many other people with rare diseases and their families, tired of a focus on what they cannot do (instead of what they can do, or the future cures they will have), and physicians talking in front of their kids in a way that doesn’t respect them. We need to change the narrative to one of hope and positivity and respect for people with rare diseases.
Arndt Rolfs, CEO of Centogene, made a similar plead to the WODC audience about the need to change the narrative. If we call them rare diseases, he explained, physicians will think that they will never across one, so they will not watch out for rare diseases. We should call them frequently misdiagnosed diseases instead, so that physicians are particularly alert to not miss them. This makes so much sense, and has such important consequences to patient diagnosis, that we should all start using the “frequently misdiagnosed diseases” term a lot more often.

With his presentation, Dr Rolds echoes Carol-Anne’s message: we need to change the narrative around rare diseases if we want to diagnose, treat and respect people with rare diseases.
5 – Reshaping “Rare” by thinking “many diseases at a time”
Chris Austin, Director of NCATS, gave one of my favorite presentations at the WODC about reshaping “rare”. At the current rate of orphan drug approval it will take 2,000 years before we have treatments for all rare diseases (assuming each new drug is approved for a different rare disease, which is not the case). A radical change in the way we discover and develop these therapies is needed. Chris’ proposal is to move from working on “one disease at a time” to working on “many diseases at a time”, exploiting commonalities among rare diseases and platform technologies to diagnose them and treat them. NCATS has multiple initiatives around this “many diseases at a time” approach, from empowering patient communities to get active in research to providing guidance on interoperable registries and partnering in drug development, including developing a gene therapy platform.
While Chris advocated for these multiplexing approaches to diagnosis and therapy development, I think that we will need some regulatory innovation to address the challenge that in some fields companies are forced to run pivotal trials in a rare disease at a time, making it not viable for companies to run trials in ultra-rare diseases and condemning these patients to off-label drug use (more thoughts on this regulatory challenge here).
6 – The place for social media and on-line patient platforms in orphan drug development
An interesting topic that also was present in different tracks at the WODC was the importance of social media and digital platforms in research and development. Luke Rosen, Head of Patient Engagement at Ovid Therapeutics, explained how Ovid believes that listening to the patient community voice is crucial for companies to understand what truly matters to families affected by rare diseases, and how Ovid is supporting the conversation by helping create community sites. A patient community that comes together becomes a stronger community, and an essential part of the drug development process. And Luke understands this well because he is also the Founder of the KIF1A.org Foundation to develop therapies for his daughter and other children with KIF1A associated neurological disorder.

We also had a workshop during the first day of the WODC, where I participated, where we reviewed how on-line communities are able to capture the patient voice in a way that is directly usable for drug development.
DuchenneXchange, for example, was created to help connect, educate and inform the Duchenne muscular dystrophy community, and James Valentine explained how regulators appreciate the use of on-line platforms to collect patient experience and preference data that will be useful for regulatory purposes. In fact in the FDA guidelines for Patient Focused Drug Development meetings, one of the methodologies for collecting patient experience data are precisely “Social Media and Identifiable Patient Communities”.
And “community” is perhaps the main value of the World Orphan Drug Congress. As much as I enjoy the workshops and presentations and roundtables, the main value I get from attending the WODC is the network. I get to catch up face to face with people I already knew, and I get to meet new people from promising companies or related patient communities.
See you all next year!
Ana Mingorance PhD