

Epilepsy Insights

MAIN LESSONS FROM THE 2025 CDKL5 FORUM
For the past eleven years the Loulou Foundation hosts an annual meeting where scientists and drug developers working on CDKL5 deficiency, together with representatives from patient organizations, meet to discuss the latest advances.
Here are the main news and take-home messages from the 2025 CDKL5 Forum that took place in October 27-28 in Boston
For the past eleven years, the Loulou Foundation has hosted an annual meeting, the CDKL5 Forum, where scientists and drug developers working on CDKL5 Deficiency Disorder (CDD), together with clinicians and representatives from patient organizations, meet to discuss the latest developments in the field and to advance towards treatments and cures. You can find summaries from some of the last meetings here: 2018, 2019, 2020, 2021, 2022, 2023 and 2024.
The 2025 CDKL5 Forum edition took place October 27-28 in Boston, and will return to London next year for the 2026 edition. This was again the largest Forum to-date, with over 200 participants including representatives from 27 companies and 22 national patient groups.
I will try to summarize the main take-home messages from this year’s Forum. This won’t cover all the presentations, so I apologize in advance to all the scientists and doctors whose work I won’t cover. My focus will be on the main themes that we saw and the main progresses towards treatments.
1. Expanding our (useful) knowledge of CDKL5

Image: Forum Director Dr Dan Lavery (Loulou Foundation); Dr Enya Paschen (Ulysses Neuroscience); Prof Peter Kind (University of Edinburgh); summary of some CDKL5 direct targets; Ulysses Neuroscience seizure platform.
Before this 2025 Forum, I thought that maybe we have uncovered most of what we need to know about how the CDKL5 kinase works, and therefore any new discovery would barely move the needle. And boy was I wrong! At this Forum, we learnt about multiple discoveries for how CDKL5 plays roles far beyond what we thought. And many of these, are useful for treatment design.
For example, we learnt about a discovery made at the same time in two countries that CDKL5 controls a protein that binds to RNA, which means that when CDKL5 is active it not only turns on and off other proteins (as kinases do), but it also controls how much of other proteins gets made. CDKL5 is also involved in autophagy, which is how cells get rid of protein aggregates. So that is another way to control how much or how little of other proteins stays in the cell. And another lab discovered how CDKL5 controls several synaptic proteins because it has a Velcro-like domain that sticks to synaptic proteins (a PDZ-binding domain). And these new functions are added to all what we knew already! like CDKL5’s control of the ion channel Cav2.3, or its modulation of microtubule dynamics and plasticity.
What this means for scientists like me who work in therapy development is an expanding map with more new targets, and therefore more therapeutic directions that we can try to pursue. So it might look like more complexity is bad, but it is actually good news. We also had a presentation from Ulysses Neurosciences showing their mouse testing platform for CDD, including behavioral readouts in male and female mice and also seizures and EEG readouts, which will make it easier to evaluate all those therapeutic options.
Bottomline: scientists are still mapping the “dark matter” of CDKL5 biology, and each of these new discoveries is a potential clue into how to develop therapies for CDD. The 2025 Forum was exceptionally fruitful on this front, with many new biology discoveries.
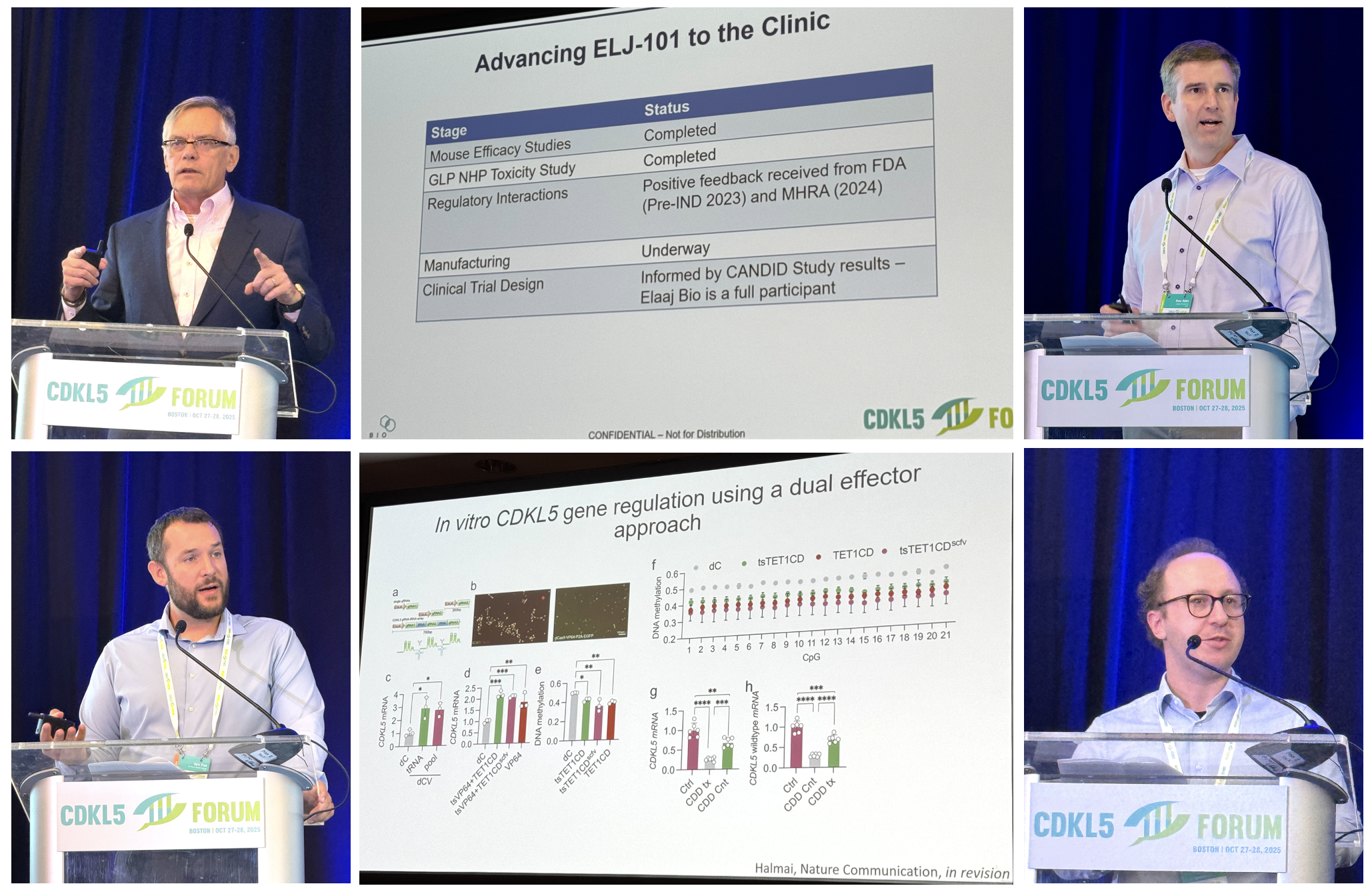
2. More options to access new medicines than ever before

Image: CDD pipeline listing most programs in development; moderator Prof Orrin Devinsky (NYU Langone); Angel Neurotherapeutics program; fenfluramine activity (UCB Pharma); Bexicaserin trial recruitment sites (Lundbeck); Praxis CSO Prof Steve Petrou.
During his introductory remarks, Dr Dan Lavery from the Loulou Foundation showed a slide of the CDD pipeline, showing multiple generations of small molecule drugs and gene therapies all in trials or advancing towards trials for CDD.
And the most important thing: in 2025 we have one drug approved, a second one having successfully completed Phase 3 trials, and two global Phase 3 trials actively recruiting patients with CDD. There was never a year before like this 2025.
This is the first year that we had Immedica at the Forum, after their acquisition of Marinus. The Immedica team presented their learnings about a slower titration with ganaxolone to reduce side effects. Ganaxolone is already commercialized in the US, and Immedica is working on market access for other countries.
This summer, UCB Pharma announced that their Phase 3 trial with fenfluramine in CDD had been positive, and at the Forum they shared the complex mechanism of action of fenfluramine, which includes more than the serotonin receptor 5HT2C. They will announce the exact results from the CDD trial at the American Epilepsy Society meeting in December, so we will watch for those. UCB will file for expanding the label of fenfluramine to also include treatment of seizures in CDD.
Last year, the regulatory agencies started allowing companies to run clinical trials combining different epilepsy syndromes and not just one at a time. This has resulted in new clinical trial options for CDD. And because these trials ask for only 4 countable seizures a month and include children as well as adults, they have given us more options than ever to try new epilepsy medicines as part of clinical trials. At the Forum we saw two large Phase 3 trials that are currently recruiting across many countries:
Lundbeck is running the DEEp OCEAN Phase 3 trial with their drug bexicaserin. Bexicaserin uses one of the receptors for fenfluramine, 5HT2C, and had very good efficacy in the Phase 2 trial also in a mix of syndromes. They have about 100 trial sites in the US, Australia, Europe and Asia. Here is the link to the study: https://deepdeestudy.com/
Praxis is running the EMERALD Phase 3 trial with their drug relutrigine. Relutrigine is a next-generation sodium channel blocker with many convenient attributes: no titration needed, once a day dosing, liquid formulation, and the trial can be done from home – they send a doctor and a nurse to your place. The trial is also in many countries, and the website is in 12 languages. Here is the link to the study: https://www.resiliencestudies.com/emerald
In addition to this first generation of treatments, all design to reduce seizure frequency, we got a glimpse for what we might have in a few years: the precision medicines for CDD. These are drugs that can correct some of the key aspects of the CDD biology. This year, we got a presentation from Dr Massimiliano Bianchi from Angel Neurotherapeutics showing their preclinical work with a drug called PME and related analogues. These drugs bind to cytoskeleton-binding proteins to compensate for the loss of CDKL5, and the most advanced drug could reach trials in CDD by 2027. This is one of multiple shots on goal to have precision medicines for CDD reach clinical trials in about two years, and because they target key aspects of the CDD biology, they are expected to also improve non-seizure symptoms as shown for the Angel lead program.
Bottomline: CDD families currently have more access to new experimental therapies than ever before. And this also includes adult patients that had not been eligible for trials until now.
3. Gene therapy progress, and a lesson in safety

Image: Keynote speaker and moderator Prof Jim Wilson (Gemma Bio); Elaaj gene therapy program (Loulou Foundation); Dr Russ Addis (Loulou Foundation); Dr Kyle Fink (UC Davis); UC Davis X reactivation project; Andrew Steinsapir (Apertura Gene Therapy)
Gene therapy remains one of the major topics at the CDKL5 Forum, and this year we received messages of excitement and also caution. Some of the Alliance members at the conference told me how “it felt like real talk”, and how that made them feel that progress in gene therapies for CDD was more tangible and real than ever before.
We learnt about progress in THREE gene therapies for CDD that are in development:
1. Dr Sharyl Fyffe-Maricich from Ultragenyx spoke about their gene therapy program, addressing “the elephant in the room” which is that they can’t give us any timelines yet for when this program might progress into trials. But she gave us a valuable scientific gift, sharing with us their research in monkeys that shows that neurons that already make CDKL5 have room to make some more CDKL5 from the gene therapy. This is important for the many CDD patients with missense mutations, who make CDKL5 but it is not functional because it carries one wrong aminoacid. Sharyl’s research indicates that they are likely to end up having a mix of CDKL5 proteins, some coming from their mutated gene and some from the gene therapy, so this is very positive. I was very relieved and grateful to learn about Ultragenyx’s findings.
2. Dr Russ Addis from the Loulou Foundation presented an update on the gene therapy that the Foundation is advancing. This gene therapy has been made in collaboration with Prof Jim Wilson, using a virus very similar to AAV9 that is now being used in 4 other clinical trials. Inside, the virus carries a copy of the CDKL5 gene and it is administered by injection into the brain CSF. Russ showed us the dose-finding studies in mice with CDD to identify the best clinical dose, and told us that the toxicology studies in monkeys have already been completed. The Loulou Foundation is now starting manufacturing, which is the large production of the gene therapy so that it can go to trials, and is already designing the clinical trial. Russ explained that manufacturing will take “the better part of a year” so we are looking into a clinical trial authorization around the end of 2026 to start trials in 2027. The Loulou Foundation announced a partnership with Gemma Bio, Jim Wilson’s company, to share data from the other gene therapies that use this same design and that are now in clinical trials. And to advance this and other types of gene therapy programs, the Loulou Foundation has created a subsidiary called Elaaj Bio, that as Russ said has a “vision for families and patients have more than one option, and to have more chances to access these therapeutics”.
3. Dr Kyle Fink from UC Davis and his lab are developing a very cool gene therapy for CDD: the X reactivation therapy. It works by using a virus to deliver to each neuron the instructions to read the second CDKL5 gene in the second X chromosome that all girls have. These instructions are a type of CRISPR. He reminded us of how nice his gene therapy works in CDD mice, and all their new progress to turn this mouse science into a human medicine. The new progress included figuring out the best doses to use, and checking that it can work in human neurons (using patient-derived organoids). They have started manufacturing and will soon start the toxicology studies in monkeys to progress into trials. This version of the gene therapy uses a combination of two virus that are administered into the CSF, but Julian Halmai (another UC Davis Professor) is working with Kyle to make a miniature CRISPR version that will enable in the future to do this with only one virus.
We also learnt about one gene therapy program that has been stopped:
This was an interesting surprise. Ashton Brennecke from Biogen explained how Biogen had been working in secret on a gene therapy for CDD, and now that the company has terminated the program they wanted to share some of their learnings with the scientific community. In their case, their gene therapy was very similar to the ones from Ultragenyx and the Loulou Foundation, and they studied how to use EEG in mice as a measure for efficacy. It was very interesting to hear about how they optimized EEG methods to be able to see the gene therapy efficacy, but it was also sad to hear that Biogen has joined Amicus and PTC Therapeutics as companies that had started gene therapies for CDD but stopped them before getting to trials. This type of attrition is common in therapy development, and the reason why we need so many shots on goal.
And importantly, we heard about what the field has learnt since the first gene therapy was tried in the first patient in the 90s:
Prof Jim Wilson is of the pioneers in gene therapy development, and delivered a spectacular presentation about the history and learnings on this field. Jim is the former Director of the Orphan Disease Center at UPenn and current CEO of Gemma Bio, the gene therapy company working with the Loulou Foundation on the CDD gene therapy. He explained how they discovered early on that to get gene therapies we would have to fight the immune system, even if it took them 17 years to understand exactly some of the key challenges. He discovered AAV9, and has now put several therapies in clinical trials both using AAV9 and his new generations of virus. Jim dedicated the last part of this talk to address a key topic right now in rare diseases: the recent cases of patients death in clinical trials with intravenous administration of high doses or AAV. His lab has studied the toxicity of gene therapies in almost 500 monkeys, and Jim asked for transparency in the field so that when a company has some unfortunate patient death in a trial, they share the data for how it happened exactly. Only that way, Jim and others can develop preclinical (animal) tests to anticipate that safety issue before it happens in a trial.
Andrew Steinsapir, from the company Apertura Gene Therapy, talked about the capsid (virus) that they are developing for neurological diseases and that is delivered by intravenous administration. The virus is engineered to cross from blood to brain by binding to receptors in the brain blood system. There are previous cases of therapies that used large proteins (although not yet virus) that can also cross from blood to brain using that same receptor, so the Apertura team believes that they should not find unexpected problems once their first programs reach clinical trials. If proven safe in trials, this type of virus could be used to create a second-generation of gene therapies for CDD that instead of being administered by a surgeon into the CSF could be simply injected intravenously.
Last, Dr Basel Assaf, an excellent toxicologist working at Attentive Science, seconded Jim Wilson’s message that so far, the field has been able to use monkeys (non-human primates) to anticipate any toxicity problem that can be later found in patients. However, this has not been perfect, and there was recently a patient death in a clinical trial in another neurological rare disease using an intravenous gene therapy that was not predicted in the monkey studies. Basel also had a great way to help us understand what we mean by “high doses”: an intravenous gene therapy can use seven quadrillion viruses, which is more than the number of cells in our body!
As a reminder: the gene therapies that Ultragenyx, the Loulou Foundation and UC Davis are developing, are NOT delivered using high dose intravenous administration. They use smaller doses, and are delivered straight to the CSF in the brain.
Bottomline: The gene therapy field is learning about safety risks, which requires transparency and collaboration. We have multiple shots on goal to have a gene therapy for CDD, with the possibility for a first trial in 2027. My overall message from this gene therapy session was quite positive. As a CDD dad put it in social media, “real change is coming”.
4. We are probably ready to run complex clinical trials

Image: Panel discussion “Where do we stand today with clinical outcome measures for CDD?”. Dr Xavier Liogier (Loulou Foundation); Dr Barry Ticho (Stoke Therapeutics); Dr Heather Olson (BCH); Dr Jenny Downs (Kids Research Institute, Perth); missing in the image Prof Tim Benke (University of Colorado); Dr Billy Dunn (Loulou Foundation, former FDA)
For the past 5 years of so, we have been hearing about developing and validating outcome measures for running clinical trials in CDD that measure symptoms beyond seizures. Looking at the 2025 Forum presentations, it looks like 80% of the work has been done, and if we had to start a clinical trial tomorrow with a gene therapy we would know what measures to include.
Dr Xavier Liogier from the Loulou Foundation presented the latest update on the international CANDID study, validating for CDD scales that have been used to measure cognition, behavior and motor skills in other diseases, and that regulators accept for drug approvals. By December of this year, CANDID will have 100 patients monitored over 2 years with this collection of scales, giving us longitudinal data for how m much change is expected over a 2-year period. Several scales and subscales are suitable for CDD, and a manuscript is under review for publication. One learning from CANDID with important trial implications is that patients with CDD have one to five seizures a day, so most will qualify for the current seizure clinical trials. This is much more than other epilepsy syndromes.
Prof Tim Benke from University of Colorado, and Dr Jenny Downs from the Kids Research Institute in Perth, presented their work to develop novel clinical scales specifically tailored for people with CDD. The clinical study that Tim leads is also close to having 100 patients reach the 2-year follow up, and importantly it also includes EEG studies so it might help identify a biomarker to see what is happening inside the brain with a gene therapy. Meanwhile Jenny is developing a novel scale for communication in CDD, which is often the top priority for families, and was praised by a former FDA Director for the very hard and diligent technical work that they are doing in developing this scale.
At a panel afterwards, presenters, including ex-FDA Director Dr Billy Dunn and Stoke Therapeutics CMO Dr Barry Ticho, highlighted that clinical trials and patient care have different goals. While patient care requires you to look at the entire complexity of the disorder, and to focus on what matters the most to every single individual patient, it is “not necessary to show everything under the sun [in trials] to get one drug approved”. Billy Dunn warned about “creating unintended obstacles” for clinical trials, and advised developers that “you don’t have to put everything in the trial and put it all in the label”. Instead, he recommended using post-approval studies to get a broader picture of the drug potential, and let doctors explore the value for each individual patient once the drug is approved.
Bottomline: There has been much progress to develop and validate clinical scales for trials in CDD beyond seizures. Because of this work, if we had to start a clinical trial tomorrow with a gene therapy we would know what measures to include. This has been possible because of hundreds of CDD families that have been participating for 2 years (some even more!) in these observational studies where they don’t get any direct benefit, only burden. You should all be proud for making this happen!
5. A strong global community and a global alliance of hope

Image: Lili Hass message and picture (CURE5); Dr Maria Luisa Tutino message; Dr Maria Luisa Tutino together with Lynn and Majid Jafar; Dr Katheryn Frame presentation and picture (CDKL5 Alliance)
The international community, and in particular the CDKL5 Patient Alliance, were repeatedly described as “a global alliance of hope”, united around people rather than around data or molecules.
Beyond just the patient groups, the collaboration among scientists, clinicians, companies, and patient advocates was credited for accelerating progress from molecular insights to therapies for CDD. As a reflection to this collaboration, there were CDKL5 Forum Awards of Excellence granted to UCB Pharma as the company making a difference, Dr Kyle Fink from UC Davis for Lab of the Year, and Dr Massimiliano Bianchi for Champion of Progress, as well as several Junior Fellowship Awards.
The Forum opened with the voice of the patient, with Lili Hass, mom of Margot and co-Founder of CURE5 sharing her family story and “seizures are just the side dish; developmental challenges are the main course”. She asks scientists to look for more therapeutic benefits and to also look beyond drugs and into tools that can make life easier for families like hers, such as equipment, feeding innovations, and better ways to track data from home.
Dr Maria Luisa Tutino, in her dual role as mum of Elettra and protein scientist, gave a talk during the gala dinner and explained that science is not just experiments, it is people, connections and persistence. Allyson Berent from the FAST Angelman Foundation echoed these words for urgency and persistence, as another rare disease mum and scientist.
And Dr Katheryn Frame, mum of Kiera, gave the last presentation, as the Chair of the CDKL5 Alliance, talking about “a world of united hope” and highlighting the work being done by so many CDD patient organizations across the globe. Katheryn explained that to her, “united in hope means strength, determination, resilience, compassion and connection”.
Majid Jafar, dad of Alia and co-Founder of the Loulou Foundation, closed the meeting reminding us that the growth of the CDKL5 Alliance is a huge part to the strength of this collaboration. As in other editions of the Forum, he reflected on how we have progressed farther and faster than we ever thought, but never as far or as fast as families would want to. Majid asked the attendees to renew our dedication to developing cures for CDD, and invited us to meet in one year for the 2026 CDKL5 Forum in London, UK.
And that’s it for the 2025 CDKL5 Forum! where we learnt about unexpected CDKL5 biology that opens new doors for treatment, saw more families eligible for clinical trials than ever before, and where the progress in gene therapies for CDD felt more tangible and real than it has ever been. This was a great Forum.
I hope you enjoyed this summary, and I’ll see you in London.
Ana Mingorance, PhD
Disclaimer: This is my own summary and key learnings, and not an official text about the Forum by the Loulou Foundation. I write these texts with the parents of people with CDD in mind, so excuse also my lack of technical accuracy in parts.

REPASO DEL FORO CDKL5 2025
La decimoprimera edición del Foro CDKL5 tuvo lugar en Boston, los días 27 y 28 de octubre de 2025. El Foro es una reunión anual que organiza la Fundación Loulou y en la que científicos y miembros de la industria farmacéutica se reúnen con representantes de la comunidad de pacientes para repasar los últimos avances en el campo.
Este es un repaso para los grupos de pacientes de las principales novedades del Foro CDKL5 2025.
Hace ya diez años que la Fundación Loulou organiza una reunión anual, el Foro CDKL5, donde los científicos de academia y de industria trabajando en el síndrome de deficiencia en CDKL5 (CDD), junto con representantes de los grupos de pacientes, se reúnen para compartir las últimas novedades y avanzar hacia tratamientos y una cura. Tenéis el resumen de los años pasados aquí: 2018, 2019, 2020, 2021, 2022, 2023 y 2024.
La edición de 2025 tuvo lugar en Boston los días 27 y 28 de octubre, y volverá a Londres para su edición de 2026. Este ha sido de nuevo el Foro más numeroso, con más de 200 participantes incluyendo representantes de 27 empresas y 22 grupos de pacientes.
Intentaré resumir las conclusiones principales de la edición de este año, no incluyendo todas las presentaciones sino centrándome en los temas principales y los progresos que vimos, que son muchos. Así que disculpas por adelantado a los científicos y médicos cuyas presentaciones no incluyo.
1. Ampliando aún más nuestro conocimiento sobre CDKL5

Fotos: Dr Dan Lavery (Fundación Loulou); Dra Enya Paschen (Ulysses Neuroscience); Prof Peter Kind (Universidad de Edimburgo); Imagen de algunas de las funciones clave de CDKL5; Resumen de pruebas de epilepsia en ratones con CDD en Ulysses Neuroscience.
Antes de este Foro 2025 yo pensaba que quizás habíamos averiguado ya todo lo principal sobre cómo funciona la quinasa CDKL5, y que por tanto los descubrimientos que faltan serían pocos y sin gran importancia. ¡Estaba equivocadísima! en este Foro oímos de varios descubrimientos que muestran que CDKL5 hace muchas más funciones que las que conocíamos, y muchas de esas son muy útiles a la hora de pensar en tratamientos.
Por ejemplo, científicos de dos países han descubierto a la vez que CDKL5 regula a una proteína que se une al RNA, lo que quiere decir que CDKL5 no solo enciende y apaga la actividad de otras proteínas (que es lo que hacen normalmente las quinasas) sino que también regula los niveles de otras proteínas. CDKL5 también estaría involucrada en autofagia, que es cómo la célula degrada agregados de proteínas. Así que de nueva esta es otra forma en la que CDKL5 estaría decidiendo lo mucho o poco que haya de otras proteínas en la célula. Y otro laboratorio ha descubierto que CDKL5 interacciona con varias proteínas sinápticas porque contiene un dominio tipo velcro que le permite pegarse a esas proteínas sinápticas (un dominio PDZ). Y esta información nueva hay que añadirla a las funciones importantes que ya sabíamos como el control de actividad del canal Cav2.3 o la modulación de los microtúbulos y la plasticidad neuronal.
Lo que esto significa, para científicos como yo que trabajamos en desarrollo de fármacos, es un mapa cada vez más amplio de posibles dianas terapéuticas. Así que aunque parezca que más complejidad es mala, en realidad es buenas noticias. También tuvimos una presentación de Ulysses Neurosciences que nos mostró su plataforma preclínica de evaluación de fármacos en ratones con CDD, en este caso tanto con ratones macho como hembras e incluyendo epilepsia y EEG. Es la plataforma más completa que existe.
En resumen: los científicos siguen descubriendo lo que se esconde en la biología de CDKL5, y cada uno de esos descubrimientos es una posible idea de cómo desarrollar tratamientos. Y el Foro 2025 estuvo particularmente lleno de nuevos descubrimientos de la biología de CDKL5.
2. Más opciones de entrar en ensayos que nunca hasta ahora

Fotos: lista de programas terapéuticos en desarrollo para CDD que presentó Dan Lavery; Prof Orrin Devinsky (Universidad de Nueva York); cómo funciona el fármaco de Angel Neurotherapeutics sobre el citoesqueleto; cómo funciona fenfluramina; lista de hospitales que tienen el ensayo clínico de bexicaserina (Lundbeck); ponente de Praxis el Dr Steve Petrou.
En su introducción, EL Dr Dan Lavery de la Fundación Loulou mostró una diapositiva de la lista de programas en desarrollo para tratar CDD, incluyendo varias generaciones de fármacos seguidos por varias generaciones de terapias génicas, todos avanzando hacia ensayos.
Y lo más importante es que en 2025 tenemos un fármaco aprobado, uno que ha completado con éxito el ensayo global de fase 3, y dos más que están reclutando ahora mismo en otras dos fases 3 globales. Jamás hubo un año con tantas opciones como 2025.
Este año por primera vez tuvimos a la empresa Immedica en el Foro, tras haber comprado a Marinus (que desarrolló la ganaxolona). El equipo de Immedica nos presentó cómo han aprendido que es mejor hacer la subida de dosis de ganaxolona más despacio que lo que pone la ficha oficial, para minimizar así los efectos secundarios. Ganaxolona ya está comercializada en EEUU y la empresa está trabajando para hacerla llegar al mercado en más países.
Este verano la empresa UCB Pharma anunciaba resultados positivos en su fase 3 de fenfluramina en CDD. En el Foro, nos explicaron en más detalle el complejo mecanismo de acción de fenfluramina, que va más allá que solo unirse al receptor de serotonina 5HT2C. Los resultados exactos de esa fase 3 nos los contarán en diciembre, en el congreso de la Sociedad Americana de Epilepsia, con lo que estaremos pendientes. Y UCB va a pedir la ampliación de la indicación terapéutica de fenfluramina para incluir también el tratamiento de las crisis en CDD.
El año pasado, las agencias regulatorias permitieron por primera vez que las empresas hicieran ensayos clínicos juntando los diferentes síndromes con epilepsia. Hasta entonces había que hacer los ensayos en cada síndrome por separado. Y esto nos ha llevado a tener más opciones de ensayos clínicos para CDD. Además, como estos ensayos incluyen niños y adultos y solo piden 4 crisis contables al mes, hay más personas con CDD que nunca antes que cualifican para estos ensayos y que pueden acceder a estos fármacos experimentales para la epilepsia. En el Foro vimos la presentación de dos ensayos globales de fase 3 que están ahora mismo reclutando en muchos países:
Lundbeck está reclutando para su ensayo de fase 3 DEEP OCEAN con bexicaserina. Bexicaserina usa uno de los receptores de fenfluramine, el 5HT2C, y tuvo resultados muy positivos en la fase 2 que también era de síndromes mezclados. Tienen unos 100 hospitales de EEUU, Australia, Asia y Europa (incluida España) en el ensayo, y el enlace de su ensayo es este: https://deepdeestudy.com/
Praxis está reclutando para su ensayo de fase 3 EMERALD con relutrigina. La relutrigina es un bloqueante de canales de sodio de última generación con muchas propiedades interesantes: no requiere ajuste de dosis (se empieza en la dosis buena), es solo una toma al día, tiene formulación líquida y para el ensayo no hace falta ir al hospital porque ellos te mandan un médico y una enfermera a casa en cada visita. El ensayo también está en muchos países (incluida España) y su web en 12 idiomas. El enlace de su ensayo es este: https://www.resiliencestudies.com/emerald
Además de esta primera generación de fármacos, que van todos dirigidos a reducir las crisis, pudimos ver un anticipo de lo que será la segura generación de fármacos: las medicinas de precisión. Las medicinas de precisión son aquellas dirigidas a aspectos clave de lo que está mal en neuronas con CDD. Las medicinas de precisión serían como un antibiótico para tratar una neumonía. Sigue siendo un fármaco al igual que el ibuprofeno, pero mientras que el ibuprofeno trata un síntoma (en este caso la fiebre), el antibiótico trata el problema de base. En el Foro vimos una presentación de Angel Neurotherapeutics, y su trabajo en ratones con CDD para desarrollar un fármaco llamado PME que corregiría el problema de microtúbulos que ocurre en ausencia de CDKL5. Y nos mostraban mejoras en los problemas de neurodesarrollo de los ratones con CDD, como no saber hacer un nido para dormir o tener problemas motores. Anticipan poder estar en ensayos en 2027.
En resumen: muchas más familias con CDD tienen opciones de entrar en ensayos que nunca hasta ahora. Y esto incluye a los pacientes adultos, que hasta ahora no entraban en casi ningún ensayo. En un par de años, esperamos empezar a tener ensayos clínicos con las medicinas de precisión.
3. Progreso en las terapias génicas, y una lección en toxicidad
Fotos: Prof Jim Wilson en su ponencia principal; diapositiva y ponente de la Fundación Loulou el Dr Russ Addis (terapia génica); ponente y diapositiva de la Universidad de California Davis el Dr Kyle Fink (terapia génica de reactivación del cromosoma X); ponente Andrew Steinsapir (Apertura Gene Therapy)
El tema de las terapias génicas sigue siendo el principal del Foro CDKL5, y este año recibimos tanto mensajes optimistas como mensajes de tener cuidado. Algunos miembros de la Alianza CDKL5 con los que hablé lo definían como que “por fin parece que vamos en serio”, y cómo este año el progreso que ha habido en el desarrollo de terapias génicas para CDD les ha hecho verlo como una realidad más tangible que nunca.
Tuvimos presentaciones de no una sino TRES de las diferentes terapias génicas que hay en desarrollo para CDD:
1. La Dra Sharyl Fyffe-Maricich de Ultragenyx habló de su terapia génica empezando por responder la gran pregunta, que es que aún no pueden decirnos cuando llegarían a ensayos clínicos. Pero nos dio un gran regalo científico al presentar su investigación en monos que muestra que las neuronas que ya tienen CDKL5 aún tienen la capacidad de producir más si se les da una terapia génica. Esto es importante para las muchas personas con CDD que tienen mutaciones missense, que son las que cambian un aminoácido de la proteína por otra, con lo que tienen CDKL5 que no funciona pero que está ahí. La investigación de Sharyl apunta a que al final se produciría una mezcla de proteínas CDKL5 en la neurona, algunas viviendo del gen que está mutado y algunas de la terapia génica, lo cual es muy buenas noticias. Así que me llevé ese alivio y gratitud a Ultragenyx por contarnos ese avance de conocimiento.
2. El Dr Russ Addis de la Fundación Loulou presentó por fin una actualización de cómo va la terapia génica para CDD que está desarrollando la Fundación. Esta terapia génica la están (estamos) haciendo en colaboración con el Profesor Jim Wilson, usando un virus que es muy parecido al AAV9 y que ya está en otros 4 ensayos clínicos ahora mismo. Y lo que lleva ese virus dentro es una copia del gen CDKL5, que se administra directo al líquido cefalorraquídeo (por la nuca). Russ nos enseñó los experimentos en ratones para dar con la dosis correcta y nos dijo que ya han terminado todos los experimentos de toxicología en primates, con lo que han comenzado la producción del virus para avanzar a ensayos. Esta parte tarda, nos dijo, “más o menos un año”, con lo que espera pedir la aprobación de ensayos a final de 2026 para empezar los tratamientos en 2027. La Fundación Loulou anunció una colaboración con la empresa Gemma Bio de Jim Wilson para compartir los datos de esos otros ensayos clínicos de terapia génica que usan un diseño muy igual a esta. Y para avanzar este y otros proyectos, la Fundación Loulou ha creado una biotecnológica llamada Elaaj Bio, propiedad única de la Fundación, con la visión de “que las familias y lo afectados tengan más de una opción terapéutica, y más opciones de acceder a todas esas terapias”.
3. El Dr Kyle Fink, de la Universidad de California Davis, está desarrollando la famosa terapia génica de reactivación del segundo cromosoma X. Esta terapia funciona también usando un virus, pero en vez de llevar el gen CDKL5 dentro lo que lleva son las instrucciones para leer la segunda copia del gen CDKL5 que todas las niñas y mujeres tenemos en nuestro segundo cromosoma X. Esto lo consigue usando una variante de CRISPR. Ya en otros años nos había mostrado lo bien que funciona esta terapia en los ratones con CDD, y este año nos vino a contar su “progreso convirtiendo la ciencia en medicina”. El nuevo progreso incluye también identificar la mejor dosis para usar, y comprobando que funciona si la neurona es humana y no de ratón, usando organoides a partir de células pluripotentes de pacientes. En este caso están comenzando los estudios de toxicidad en monos y la producción del virus para ensayos todo en paralelo, porque ya la terapia génica está optimizada tal cual está. No dio fechas, pero si que nos explicó que su colega el Dr Julian Halmai está trabajando en una variante de CRISPR en miniatura que les permita meter toda esta terapia en un virus solo, ya que actualmente utilizaría dos tipos de virus (uno con la primera mitad del CRISPR y otro con la segunda, que luego se encuentran en cada neurona).
También pudimos oír de una terapia génica que ha sido parada:
Esta fue una sorpresa interesante. Ashron Brennecke de Biogen, subió al pódium para explicarnos que Biogen había estado trabajando también en una terapia génica para CDD en secreto, pero que habían decidido eliminar el programa. Ahora que ya no están trabajando en CDD, querían compartir con nosotros el método para medir eficacia de la terapia en ratones usando EEG y que creen que se podría usar también en ensayos clínicos. Estuvo interesante oír ese trabajo, pero a la vez es bastante triste saber que ya van tres, y que Biogen se une a Amicus y a PTC Therapeutics como empresas que empezaron pero luego abandonaron el desarrollo de terapias génicas para CDD. Pero al fin y al cabo esto es ciencia y esto son empresas, con lo que abandonar proyectos por el camino está a la orden del día y es justamente la razón por la que debemos tener varias opciones en desarrollo en todo momento.
Y un tema muy importante, pudimos aprender de las lecciones que nos ha dejado el campo de la terapia génica desde que la primera persona recibiera la primera terapia en los años 90:
El Profesor Jim Wilson ha sido y es uno de los pioneros del desarrollo de terapias génicas, y nos dio una ponencia espectacular sobre lo que han sido estos 30 años y lo que han aprendido. Jim es el antiguo Director del Orphan Disease Center de la Universidad de Pensilvania, y el actual CEO de Gemma Bio – la empresa de terapias génicas que está trabajando con la Fundación Loulou en la terapia para CDD. Jim nos explicó cómo el primer problema que afrontaron fue que para conseguir terapias génicas tendrían que pelearse con el sistema inmune, y que les llevó 17 años (que se dice pronto) entender bien qué había pasado en los primeros casos de fallecimientos por terapias génicas. Jim es el científico que descubrió AAV9, el virus más famoso, y ya ha llevado a ensayos muchas terapias génicas usando el AAV9 o su nueva generación de virus. Jim dedicó la última parte de su ponencia a un tema del que se habla mucho ahora en enfermedades raras: los recientes casos de fallecimientos como consecuencia de la administración intravenosa de AAV en dosis altas. Su laboratorio lleva estudiando la toxicidad de las terapias génicas en caso 500 monos, y lo que Jim pide es transparencia a todas las empresas trabajando en terapias génicas para que cuando ocurra un caso de fallecimiento en un ensayo compartan con la comunidad científica exactamente qué pasó. Porque solo así podrán Jim y otros científicos desarrollar medidas para ver esa toxicidad en los estudios en monos previos a ensayos, y evitar así más fallecimientos.
Andrew Steinsapir, de la empresa Apertura, nos habló de su nuevo virus diseñado para ser administrado por vía intravenosa y que cruce la barrera hematoencefálica aprovechando unos receptores de la superficie vascular cerebral. Ya existen otras terapias que usan macromoléculas (aunque no virus) que pasan de sangre a cerebro usando esos mismos receptores, con lo que el equipo de Apertura considera que su virus no tendrá problemas de seguridad cuando llegue a ensayos. Y la verdad es que si este tipo de virus resulta ser seguro en ensayos clínicos nos permitiría tener una segunda generación de terapias génicas para CDD que en vez de requerir a un neurocirujano para inyectarla en el líquido cefalorraquídeo a través de la nuca solo requiera una infusión intravenosa.
Y por último el Dr Basel Assaf, un toxicólogo brillante que trabaja para la empresa Attentive Science, se hacía eco de las palabras de Jim diciendo que hasta ahora hemos podido anticipar en monos cualquier problema que luego apareció en la clínica. Sin embargo esta predicción no ha sido perfecta, porque recientemente nos hemos llevado una muy mala sorpresa con una terapia génica para otros síndrome de desarrollo en la que tuvieron una muerte en el ensayo mientras que los monos no habían dado ninguna señal. Era de nuevo un caso de dosis alta intravenosa, y Basel nos ayudó a hacernos una imagen mental de qué queremos decir con dosis alta de virus: una infusión intravenosa de terapia génica a dosis altas usa siete cuatrillones de virus, que son más que todas las células de nuestro cuerpo.
Por cierto que os recuerdo que las terapias génicas para CDD de Ultragenyx, la Fundación Loulou y la Universidad de California Davis, no usan ni dosis altas ni vía intravenosa. Usan todas dosis más bajas y van directas al líquido cefalorraquídeo, no por via venosa.
En resumen: la medicina está aprendiendo cómo funcionan los riesgos de las terapias génicas, lo cual requiere transparencia y colaboración. Tenemos varias terapias génicas en desarrollo para CDD con la posibilidad de ensayos clínicos en 2027. El mensaje general de la sesión del Foro sobre terapias génicas fue bastante positivo. Como un papá de CDKL5 puso en redes sociales, “se viene un cambio importante".
4. Posiblemente ya estemos listos para hacer ensayos clínicos más complejos

Foto: el panel de la sesión de escalas clínicas, con el Dr Xavier Ligioer (Fundación Loulou), el Dr Barry Ticho (Stoke Therapeutics), la Dra Heather Olson (Hospital Infantil de Boston), la Dra Jenny Downs (Kids Research Institute en Australia), falta en la imagen el Prof Tim Benke (Universidad de Colorado) y por último el ex-Director de neurología de la FDA actualmente con la Fundación Loulou el Dr Billy Dunn.
Durante los últimos 5 años o así hemos estado hablando de la necesidad de estar preparados para ensayos clínicos en CDD que miren más que las crisis epilépticas. Y cómo para eso hay que tener escalas clínicas aprobadas por reguladores y validadas para medir los otros aspectos principales de la enfermedad. Tras ver las presentaciones del Foro de este año yo creo que ya estamos listos, y que si tuviéramos que empezar mañana un ensayo clínico con una terapia génica sabríamos qué medidas usar.
El Dr Xavier Liogier de la Fundación Loulou presentó las últimas noticias del estudio observacional CANDID, en el cual en España tenemos muchas familias participando. CANDID usa escalas clínicas que ya se han usado para aprobaciones de tratamientos en otras enfermedades y que cubren los aspectos principales de CDD también. Se espera que en diciembre de este año lleguen 100 pacientes a la visita d ellos 2 años del estudio, y ya sabemos cuáles de las escalas funcionarían en ensayos y hay un manuscrito científico enviado a publicación. Una de las lecciones de CANDID, con implicación para ensayos clínicos, es que de promedio las personas con CDD tienen entre 1 y 5 crisis epilépticas contables al día. ¡Al día! con lo que la inmensa mayoría podrían participar en los ensayos clínicos que hay abiertos en este momento, porque es mucho más que en otros síndromes con epilepsia.
El Profesor Tim Benke, de la Universidad de Colorado, y la Dra Jenny Downs del Kids Research Institute en Australia, presentaron su trabajo para desarrollar nuevas escalas clínicas, específicamente diseñadas para CDD. El estudio observacional que lidera Tim también está a punto de llegar a 100 participantes con seguimiento de 2 años, y este estudio además incluye medidas de EEG con lo que podría identificar una medida electrofisiológica para usar en ensayos clínicos y ver qué está cambiando en el cerebro más directamente. Jenny está desarrollando una escala específicamente para medir la comunicación en CDD, que es muy muy importante para las familias, y que recibió los elogios de un antiguo director de la FDA por el trabajo tan detallista y diligente que están haciendo con esta escala.
En un panel de debate que cerró esta sesión, los panelistas, incluyendo ese antiguo director de la FDA el Dr Billy Dunn y el Director Médico de la empresa Stoke Therapeutics Dr Barry Ticho, señalaron que los ensayos clínicos y el cuidado médico de los pacientes tienen diferentes objetivos. Si bien durante el cuidado médico de un paciente hay que mirar a todos los síntomas y a los que más impactan a ese paciente en concreto, “no hay que mostrar todo lo habido y por haber en un ensayo para obtener una aprobación”. Billy advertía de evitar esto para “no crear obstáculos sin querer” que pudieran complicar los ensayos clínicos. En vez de sobrecargar los ensayos, recomendaba que una vez esté el fármaco aprobado ya los médicos miren todos los escenarios en los que podría funcionar ese fármaco, además del beneficio para cada paciente de forma individual. Pero todo eso una vez están ya aprobados.
En resumen: Hubo mucho apoyo desde la industria y profesionales reguladores al trabajo que llevan haciendo estos equipos para desarrollar y validar escalas clínicas para CDD que midan más allá de las crisis. Y estos estudios son solamente posibles gracias a la generosidad de cientos de familias que llevan asistiendo dos años (o algunas más) a las visitas de estos estudios, donde no reciben ningún tipo de fármaco. ¡Muchísimas gracias a todos los que hacéis esto posible!
5. Una comunidad global, y una alianza de esperanza

Fotos: Mensaje y ponencia de Lili Hass (CURE5); mensaje de la Dra Maria Luisa Tutino; foto de Maria Luisa con Lynn y Majid Jafar; mensaje y foto de la Dra Katheryn Frame (Alianza CDKL5).
Durante el Foro, varias veces se refirieron a la comunidad de pacientes internacional, y sobre todo a los países que son miembros de la Alianza, como “una alianza global de esperanza”, unida en torno a las personas más que en torno a proyectos o moléculas.
Más allá de los grupos de pacientes, la colaboración entre científicos, médicos, empresas y representantes de familias es la que ha conseguido acelerar el progreso de los descubrimientos científicos a tratamientos para CDD. Reflejando esta colaboración, en el Foro se dieron premios a la empresa UCB Pharma como empresa que ha marcado una diferencia en CDD, al Dr Kyle Fink como laboratorio del año, y al Dr Massimiliano Bianchi como promotor de progreso, todos por sus contribuciones recientes a la enfermedad. También se dieron premios a jóvenes científicos que están realizando sus proyectos en CDD.
EL Foro siempre abre con la voz del paciente, en este caso con Lili Hass, la mamá de Margot y cofundadora de CURE5. Lili explicó a la sala que “las crisis son solo el aperitivo en CDD, el plato fuerte son los problemas de neurodesarrollo”, y pidió a los científicos que busquen soluciones más allá que con fármacos, por ejemplo con equipamiento, facilitar la alimentación o facilitar la captura de datos desde casa.
La Dra Maria Luisa Tutino, mamá de Elettra y científica, nos dio una presentación durante la cena de gala explicando que la ciencia no son experimentos: es gente, es conexiones, y es perseverancia. Recibimos un mensaje similar de Allyson Berent de la fundación de síndrome de Angelman FAST, que es también madre y científico.
La Dra Katheryn Frame, mdare de Kiera, dio la última ponencia, en capacidad de presidenta de la Alianza Internacional CDKL5. Su mensaje fue de “un planeta unido en la esperanza” y destacó la labor de muchos de los grupos que son miembros de la alianza. Katheryn explicó que para ella “unidos en la esperanza significa fuerza, determinación, resiliencia, compasión y conexión”.
Majid Jafar, el padre de Alia y co-Fundador de la Fundación Loulou, cerraba el congreso recordándonos que el crecimiento de la Alianza CDKL5 es el elemento central que da fuerza a esta colaboración entre científicos, médicos y familias. Como en ediciones anteriores del Foro, hacía la reflexión de que hemos llegado más lejos y más rápido de lo que pensábamos, pero nunca es suficientemente lejos ni suficientemente rápido para lo que querrían las familias. Majid pidió a los asistentes renovar su compromiso para desarrollar curas para CDD, y nos invitó a reunirnos de nuevo en un año para el Foro 2026, esta vez en Londres.
¡Y hasta aquí llegó este resumen del Foro CDKL5 de 2025! donde aprendimos de biología que no podíamos imaginar y que abre puertas a nuevos tratamientos, supimos que había más familias con posibilidades de entrar en ensayos que nunca antes, y donde el progreso en las terapias génicas para CDD se volvió más tangible e inmediato que nunca. El de 2025 ha sido muy buen Foro
Espero que os haya gustado este resumen, y nos vemos en Londres.
Ana Mingorance, PhD
Nota: este texto captura mis impresiones de las presentaciones del Foro que más me interesaron, no es un texto oficial del congreso emitido por la Fundación Loulou. Escribo estos resúmenes para los padres de personas con CDD, así que a veces me tomo ciertas licencias a la hora de explicar las partes mas técnicas.

CDKL5 ALL IN(VOLVED) 2025
This is a scientific summary of the 2025 CDKL5 Alliance patient conference celebrated in Rome. The CDKL5 Alliance is the umbrella organization bringing together national CDKL5 deficiency patient associations as well as affiliated organizations like the Loulou Foundation. The summary covers the scientific updates presented during the conference in June 2025.
1. Intro: Back in Rome
The first CDKL5 conference that I attended happened to be the patient meeting in Rome in 2017, where the CDKL5 Alliance was started. I am a neuroscientist and I was already working with patient foundations for related disorders, so I wanted to learn about CDD. I remember meeting some Spanish families (my country) who quickly explained me the disease. Also seeing Marinus on stage talking about a very small trial that they were running in CDD with their experimental drug ganaxolone. And I remember Antonino Caridi, a CDD grandpa, who took the initial leadership of the patient Alliance and welcomed us from stage.
I ended up joining the Loulou Foundation in 2018, so having the CDKL5 Alliance meeting return to Rome in 2025 felt very special. We were back in the same place, but everything about CDD is now immensely larger: the size of the community, the treatment pipeline, and the number of people working on it. And Antonino still looks the same.
The ALL IN(VOLVED) conference started Friday June 27th, with families catching up with each other, and meetings of the national patient group leaders and also with the pharma representatives. To give you some numbers, we had over 300 participants, with 41 people with CDD and many siblings who made the conference very enjoyable. In total we had participants from 24 countries or territories! in alphabetic order: Albania, Australia, Austria, Belgium, Brazil, Bulgaria, France, Germany, India, Ireland, Italy, Japan, MENA, Netherlands, Philippines, Poland, Romania, Slovakia, Spain, Sweden, Switzerland, the UK, Ukraine and the USA. That’s why we call it the Global Alliance. And we had 9 pharmaceutical and biotech companies supporting the meeting. Just hearing these numbers would have blown the minds of the 2017 participants.
Then on Saturday 28th we started with a series of science talks for all attendees, where we reviewed the disease, the natural history studies, the pipeline, and learnt about different therapeutic modalities and where they are. We called these the ALL IN(FORMED) sessions.
We also had a presentation from the team at the Pediatric Therapy Center High Hopes, in Dubai, because the non-pharmacological therapies are so far the best therapies that we have for kids and adults with CDD. They showed us best practices to improve strength and mobility and promote neuroplasticity in these kids, and talked about a “multidisciplinary team with a family-centric approach” with parents as co-therapists.
For the afternoon we split into two tracks. I stayed with the scientists and the pharma companies in a partnering session where we learnt about specific programs on new therapies and model systems for the disease. I will mention these in my summaries below but without much detail, given that this track was designed to be only for scientists and not open to families. The parallel track for families included biomarker sampling and pet therapy with a dozen of beautiful dogs, which made us, scientists, very jealous.
And we closed the conference on Sunday June 29th with the ALL IN 1 COMMUNITY session where the families took the stage and we all listened.
I will summarize below the main news from the scientific sessions, and some of the main messages from the families that I found particularly impactful. Please keep in mind that I am looking at the therapeutic progress as a scientist, not as a parent. And this is not the entire summary of the ALL IN(VOLVED) CDKL5 Alliance conference, so apologies in advance for the parts that I might be missing.
2. Science: small molecule drugs
The first generation of treatments developed for CDD are anti-seizure drugs. That’s because CDD is one of the genetic syndromes that comes with the highest seizure frequency, and there is no good drug to control those seizures, so we really need something better.
In 2017 in Rome, Marinus presented data on 4 patients with CDD taking ganaxolone, FOUR. Three out of four had improved, the fourth one didn’t, but it looked promising. Their plan was to recruit more patients.
By the time we met back in Rome for the ALL IN(VOLVED) 2025 conference, we know that the Phase 2 trial included 7 patients and was successful, that it would be followed by a global Phase 3 trial with more than 100 patients also successful, that then Marinus succeeded at getting ganaxolone approved for treating seizures in CDD in the US and Europe, and that Marinus was later acquired by a larger company called Immedica Pharma who was now with us in Rome at the conference. Change and progress can seem slow looking forward, but quite fast when you look backwards.
Carol-Anne Partridge is a CDD mum (and leader of the UK patient group) and she presented a study designed to convey better to governments and payers the severity of CDD. This is very important because ganaxolone is the first drug approved for CDD, so when authorities evaluate their reimbursement decision on ganaxolone they are learning about the drug AND the disease at the same time. And because of this learning curve, ganaxolone is still only commercially available in the US. The study surveyed 132 families, and showed that 96.7% of patients are taking anti-seizure drugs, and that if you measure the health-related quality of life of the CDD patients as reported by their parents from 0 (a state comparable to being dead) to 1 (representing full health), the average is 0.18. The survey used the methodology that payers want to see, and was designed to “speak” to them. I think the message is loud and clear about the need for new medications.
The biggest news of this conference came from our second Phase 3 trial in CDD: UCB Pharma issued a press released the first day of the conference announcing that the Phase 3 trial with fenfluramine for treating seizures in CDD was successful, both in efficacy and safety, and that they will be moving forward to request an approval for CDD now (fenfluramine is already approved for two other syndromes). If everything goes well, this will become the second drug approved for CDD. And thank you UCB for the perfect timing with the announcement, we all got to celebrate together.

And what comes after two global Phase 3 trials? another global Phase 3 trial. The company Longboard (part of Lundbeck after an acquisition) is developing a second-generation fenfluramine, called bexicaserin, that is already in Phase 3 trials in the US and is adding many more countries world-wide progressively this year. Interestingly this drug is bringing together into the same trial all of the rare epilepsy syndromes, that are often called “DEEs”, and because people with CDD often have many seizures it means that many will qualify for this trial. It asks for a minimum of 4 countable seizures a month, and ages 2 to 65. In Rome, we had Dr Marina Trivisano present an update on this program and show us the map with all the countries in the trial. You can see more information online if you look for the “DEEp OCEAN” trial, and I encourage you to ask your neurologist about it if you are interested.
These first three large trials were for seizure medications, but I also explained in my presentation about the CDD pipeline that after these trials we are moving into therapies that treat the disease, not the symptom. And this includes not only the gene therapies (more about this in a minute) but also small molecule drugs that act on the most important targets of CDKL5. What we call “small molecule drugs” are the regular medicines that are taken orally like a pill or a syrup and that are made of a chemical compound – not something big like a virus or a protein. And because the work of the CDKL5 protein in the cell is to turn on or off other proteins, acting like a switch (that’s what kinases do), it is possible for scientists to design small molecule drugs that can bind to those proteins now, and turn them on or off as they needed.
In the morning session I introduced that concept, and I called it precision medicine because those drugs are designed to correct some of the key targets of CDKL5. And later in the afternoon during the partnering session we had a presentation from Prof. Massimiliano Bianchi who is developing one of those precision medicine approaches, with drugs that target the cytoskeleton (more of why that’s important for CDKL5 HERE in section 1).
3. Science: gene therapies
There are several gene therapies in development to treat CDD, and we hope that they will get to clinical trials in the near future. A gene therapy uses a virus to deliver a copy of the CDKL5 gene to the brain, so it is made of the outside of a virus (to help it get into brain cells) but inside it doesn’t have virus DNA, it has the CDKL5 gene.
Dr. Stuart Cobb is one of the inventors of a gene therapy for Rett syndrome (with the gene MECP2) and gave us a beautiful lecture on how gene therapies work. In his words: “it is an easy concept, but with complicated execution”. It essentially follows four steps: make the virus, cure a mouse, check for toxicity in monkeys, and then go to clinical trials. He explained how in Rett syndrome they are already in clinical trials, and they are seeing clinical improvements. And two days after his talk, the company developing his gene therapy announced that they are progressing to Phase 3 trials with their gene therapy in Rett syndrome.

Some years ago ,we thought that gene therapies for CDD would be in clinical trials by now. Ultragenyx had said at the 2022 CDKL5 Forum that they hoped to be in trials in the following year. But that hasn’t happened yet, two and a half years later, so I know that has disappointed many families who have been counting how old their kids will be by the time the gene therapies arrive to trials. And that premature announcement has probably also made the other gene therapy developers refrain from speaking too soon about their timelines, to not risk disappointing the community once again. As a result, we are still a bit in the dark about how many gene therapies are being developed for CDD and how far along they are.
Majid Jafar and I (both from the Loulou Foundation) mentioned in our presentations that there are multiple gene therapies in development for CDD, but it would have been much better to get some of the companies present an update. Even without stating timelines, seeing the progress that they are making would have been and influx of air for the 300+ family members in the room.
We did have two presentations dedicated to a special type of gene therapy for CDD: a gene therapy designed to open up the second CDKL5 gene copy. For background: because males only have one X chromosome, female cells only use one of our X chromosomes and turn the second one into a tiny little ball that is not read. That way, both males and females have one functional X chromosome. Which X chromosome gets inactivated is totally random and is different for each cell. What this gene therapy does is to carry inside of a virus the instructions to read the second copy of CDKL5, the one in the inactive X chromosome. And because girls with CDD have always one good CDKL5 gene copy (the other one is mutated) this gene therapy can help each of their cells read the two copies, one of which is good, and that’s all we always needed! Dr. Kyle Fink is leading this program and told us that they are now making “clinical grade gene therapy” to start the safety experiments in monkeys that come prior to trials.
4. Science: Enzyme Replacement Therapies
We also had a lecture on how enzyme replacement therapies work. Prof. Elisabetta Ciani explained very nicely how an enzyme replacement therapy consists of making “lab-made CDKL5 protein” to then give it to neurons. This is a good idea because CDKL5 is an enzyme, and other brain enzymes have been successfully adapted to enzyme replacement therapies. But as Stuart had said earlier that morning, this is also “an easy concept, but with complicated execution”. Elisabetta had succeeded at making a modified CDKL5 (so that it enters cells) in her lab and using it to treat CDD mice, but the same methods that worked for mice don’t work as a medicine for people, and here is where the challenge has been.
Prof Maria Luisa Tutino, who is both a CDD mum and a protein scientist, has been trying to make as many changes as needed to the CDKL5 protein to make it suitable as a lab-made protein therapy. The first-generation lab-made CDKL5 only goes to the “stomach” of neurons, so they eat it instead of using it, and she is currently working on a second-generation approach for which she has received a 1 million Euro grant. This one is clearly a very complicated therapeutic approach, but one worth fighting for.
5. Making it easier to develop treatments and to run trials
Before getting to trials, we need to believe that the treatment is going to work. How do we check that it could work? by testing it first in animals with the disease. And once we get to trials, we need to confirm that the treatment works, this time in patients. And how do we see if it worked in patients? by measuring clinical changes.
At the 2025 patient conference we got updates on animal models and on clinical change measures.
We reviewed the animal models in the partnering session, with Prof. Leonor Cancela showing us the zebrafish model of CDD (and small molecule screenings that she is running), María del Carmen Martín showing us the drosophila model of CDD (which has lots of seizures) and the Ulysses Neuroscience team showing us their CDD mouse model testing service. That means we have the entire toolbox to run preclinical trials in CDD animals.

And we got updates on the studies to measure clinical changes during the big morning session with all families in the audience, because all CDD families are critical players in these clinical programs.
Prof. Tim Benke gave us an update about the disease, and also updated us about the ICCRN natural history study in the US where they are developing a single scale to measure the global severity of CDD for each patient. And Dr. Xavier Liogier told us about the international CANDID study that is managed by the Loulou Foundation, where we have been able to see that there are many seizures at all ages, that most of the developmental improvements are seen in children under the age of 6, and that there are several scales to measure different disease domains (like cognition, communication, behavior or motor skills) that could be already used in trials. These two studies have enrolled more than 200 patients together, and participating in these studies means several hospitals visits over several years, so it is a big effort by the patients and their families and it would be impossible to design complex clinical trials (like for gene therapies) without all of you.
While the natural history studies help us know how to measure EXTERNALLY how a medicine might be helping, we use biomarkers to measure the INTERNAL changes. At the Rome conference we saw an update on the ELPIS global biomarker study by Prof. Massimiliano Bianchi, where they track changes in proteins in blood that tell us what is happening in the brain. And we also had a presentation by Vita Cardinale, who won the conference poster competition and was invited to give a talk on her project where they look at biomarkers in saliva from patients. These biomarker studies are made possible by more than 100 volunteer patients and their families who are helping these scientists identify the internal measures of change.
By the way you might wonder how blood or saliva can tell us what is happening in the brain. The answer is that neurons talk to each other by sending out little bits of neuron that contain proteins and RNAs almost like sending a letter by post to each other. The inside of those little bits tells the recipient how that original neuron was feeling. Sometimes the recipient is a neighbor neuron, but sometimes those letters travel so far that the recipient is a scientist that can pick it up in liquids like blood or saliva and read that letter. Science can be very cool.
6. Voice of the community
Patient conferences can be very emotional, with many highs and lows. In Rome we shared three days with families in very different situations, from those who had their little kids with CDD taking their first steps, to parents who have lost their children. So it was only fair that after the main conference day families took the stage.

During this final session we listed to #1minuteofhope videos where parents from all over the world told us what hope meant for them, and asked the audience “what does hope mean for you?”. We had videos from Italy, Peru, USA, Canada, Japan, Ukraine, MENA, and France.
Mais Kanan from CDKL5 MENA started the day, and Majid Jafar from the Loulou Foundation gave us a recap for how much progress has happened since his first daughter Alia got diagnosed with CDD in 2014. His was a message of hope, hard collective work that got us to where we are, and perseverance to get where we need to be.
We then got to hear from two siblings, starting with Iman Jafar who told us about how much she loves having Alia as her older sister, and how the first word that she ever spoke was “up” after hearing so often everyone tell “up!” to Alia to help her with her motor skills. And later we heard from Alessandro Caridi, who had a harder story as a sibling to a sister with CDD, but who also reminds us that the experience of every family and every person is unique, and to not underestimate the impact of this severe disorder on everyone around.
Dr Michela Fagiolini spoke about the possible future and dreams, from her experience working as a scientist in CDD and related diseases. She had some really good messages for families, like “drugs tailored for CDD and gene therapy are coming. These are not just abstract hopes, they are real and moving forward”. And how “it cannot be one person or one lab or one family, everyone is needed, and you are not alone”.
Then CDKL5 Spain shared two voices, one from a founder parent, and one from a more recently diagnosed family. Sandra Pérez spoke about being one of the “5 families and 5 stories” who created the Spanish association in 2014, and how one of the main missions of the group is and was to welcome new families, because they know how it feels to receive a diagnosis that changes everything, and how important to know that you are not alone. Then Manuel Vigara spoke about how thankful he was to those first 5 families who were “a light in the middle of dark, and whose steps opened our path”. He explained that the word “rare” often means no resources and no answers, and asked for the science community to keep dreaming and progressing, because “each hour in the lab is a thread of hope for millions of families” and “each small progress is a miracle in the making”.
We heard from the Fondazione Telethon in Italy about their support to rare genetic patient groups, and how “tenacity” is the identity of the Italian association and host of this meeting, CDKL5 Insieme Verso la Cura.
Last Barbara Verdirame, president of CDKL5 Insieme Verso la Cura, took the stage to thank everybody, from the families to the volunteers to the scientists and the doctors and the industry representatives in the room and everyone who supported the conference. Because in the end it is only through that massive collaborative effort that we get to advance medicine.
And I want to close this summary by capturing some words from Antonino Caridi, who opened the conference by reminding us of how far the International CDKL5 Alliance has come since it was founded in Rome in 2017, how “hope” is not a passive word, but one that gives us the courage to transform the injustice of CDD, and that “the best is yet to come”.
Ciao a tutti!
Ana Mingorance, PhD
Disclaimer: I write these texts with the parents of people with rare epilepsy syndromes in mind, so excuse also my lack of technical accuracy in parts. And credit for the beautiful pictures goes to Massimiliano Marcoccia and the great team from CDHL5 Insieme verso la cura.

AES 2024: THE FUTURE IS NOW
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. These are my main insights from the American Epilepsy Society 2024 meeting.
I often write a summary of the main lessons from the American Epilepsy Society meeting, but this year there was so much about epilepsy syndromes (which you also see referred to as Developmental and Epileptic Encephalopathies, or DEEs) that I didn’t have a chance to see many of the more general epilepsy presentations at AES, or even everything about DEEs!
Therefore this summary is not intended to be a summary of what was presented at AES, but simply the highlights of what caught my eye at the conference and where I see the field going.
You can see my summaries from previous years at AES here: 2017, 2018, 2019, 2020, 2021, 2022, and 2023.
1 - THE FUTURE IS NOW: TANGIBLE PROGRESS IN DISEASE-MODIFICATION
In 2022 I talked about crossing the line from developing treatments for symptoms, to developing treatments for the cause of the disease. And the line from focusing on a few syndromes only (those with the most patients), to seeing treatments in development for many more. I called that escape velocity.
At that time in 2022, that progress was illustrated by ongoing trials for Dravet syndrome (SCN1A haploinsufficiency) and preclinical progress in other DEEs. We were not there yet, we were just taking off.
After a slower 2023, 2024 caught up with the speed and delivered us a fantastic year for DEEs, and in particular for Dravet syndrome. This was the first classic DEE (if we don’t count other neurodevelopmental disorders like Angelman syndrome and Rett syndrome, more on this below) to cross the line between having promising treatments for the cause of the disease in development, to having clinical proof of disease-modification.
For Dravet syndrome, 2024 has marked a before-and-after in therapy development, and two news at AES made all this difference.
First Encoded Therapeutics presented during the Dravet Syndrome Foundation Roundtable the evening before AES and announced that they have already dosed 5 patients with their gene therapy ETX101 using a virus to deliver a transcriptional activator for SCN1A. They expect to be able to share some safety and efficacy data in the second half of next year. Encoded is only recruiting very young children for their POLARIS Phase 1/2 program in Australia, the US and the UK, as young as 6 months of age. This means that if this gene therapy is successful, these might be the first few kids with Dravet syndrome that will be spared from developing the disorder, or that will only develop a milder form of it. And as a reminder, Dravet syndrome like all other DEEs comes with neurocognitive delay, behavioral problems, motor problems, drug-refractory epilepsy and (in this case) a high rate of seizure-related mortality. Because it can often be diagnosed during the first year of life, a gene therapy like this one could be used in the future soon after diagnosis and PREVENT the development of Dravet syndrome. At least that would be the ultimate success scenario.
But what if you already developed Dravet syndrome? That was the second big news at the conference, and for me the highlight of the entire meeting and of my 13 years working in this disease. A few days after the announcement from Encoded, Stoke Therapeutics showed the complete results from their Phase 2 in children and teenagers ages 2 to 18 with zorevunersen (STK-001), an ASO designed to increase the levels of productive SCN1A mRNA, including 24 months follow up of these patients, and we learnt that:
Stoke already identified the right dosing for the Phase 3 trials: 2 or 3 loading doses of 70mg followed by 45mg maintenance doses every 4 months.
They see sustained seizure reduction of over 80% at that optimal dose (this is more than fenfluramine, and with half of the patients already on fenfluramine!)
They documented growing improvements after 12 months and 24 months in …wait for it: receptive communication, expressive communication, personal skills, interpersonal relationships, play and leisure, coping skills, gross motor and fine motor skills. That’s it, the entire Vineland-3 scale improved in all domains, getting even better into the second year of treatment, while we know from the natural history studies that these patients don’t make improvements in any of these skills within a year.
To better understand the transformational improvement that these numbers meant, one of the principal investigators from the Phase 2 trial showed some videos of the before and after of a 12-year-old treated with zorevunersen. In my experience, what I saw is what many Dravet syndrome families have described over the years as “the cure”, and what we can medically call a truly life-transforming improvement across all the disease domains. Here is some description for what we saw in the video:
Before treatment the teenage girl had problems with motor coordination and balance, hesitation in movements when trying to kick a soccer ball and ataxia. She looked like many people with Dravet syndrome at her age. Yet the video of her playing soccer 8 months after treatment with zorevunersen showed a completely different posture, much more fluid, with balance, even able to control the ball with her foot to center it before kicking. This level of improvement in this short amount of time was not something I thought would be possible, and that’s the power of seeing videos. And then again, remember that patients on zorevunersen keep getting better over time and this improvement was already within the first year.
We also saw videos of her buttoning her shirt with much improved fine motor skills, and then some videos of her talking to her doctor and following some basic directions that showed a dramatic difference between a shy distracted kid who could not keep much eye contact or follow basic instructions as in “touch my finger and then touch your nose” to a completely transformed teenager within a year, who would sit up and cheerfully follow the instructions and chat with her doctor about her school and favorite subject. And she went from 5 seizures a month to 1 or 2 seizures a month so this improvement is clearly related to the genetic upregulation and not to any dramatic effect secondary to seizure reduction. The transformation is hard to explain with words so I hope you can see the video in Stoke’s website (during this first month you can see it here) and how she does not look like the same kid after 8-12 months of treatment.
Stoke said that the improvements that we saw in this girl map into the Vineland as an average patient after treatment with zorevunersen. So this was not a super-responder case, this was a representative patient.
This clinical trial with zorevunersen in Dravet syndrome is the first proof of disease-modification in a DEE. The first successful transition from seizure management to SYNDROME management, by targeting the root cause of the disorder. Any improvements in cognition or behavior (mainly focus) that we have seen over the years with seizure drugs like fenfluramine dwarf in comparison to the magnitude and sustained progress that we are now seeing with zorevunersen. Finally the future is now.
The results with zorevunersen in Dravet syndrome are so strong that I worry about the possibility of unblinding during the Phase 3, because presumably (the design has not yet been announced) the Phase 3 will be about one year long yet clinicians from the Phase 2 report clear improvements as soon as 6 weeks into the trial. Unblinding due to efficacy already happened in the spinraza trials in SMA, and indeed zorevunersen is looking like a spinraza in the making.
And now that we know what the human brain can do, and how much plasticity is still in it if only we can come with the right genetically-targeted treatment, we look at the other DEEs and ask when will they get their Stoke moment.
For SCN2A developmental and epileptic encephalopathy, Praxis is developing an ASO to downregulate the ion channel expression in cases of gain-of-function mutations (elsunersen), but it is so far only used in a few cases under emergency use. My eyes are on the gene therapy from Capsida for STXBP1-related disorders, which is on track to get an IND in the first half of 2025 and it will be an intravenous-administered gene therapy to restore STXBP1 levels, so it is a first-of-a-kind for the DEE world and even for the entire neurology field. SYNGAP1 held a scientific conference the day before AES and they also seem to be next in line for disease-modifying trials, with the ASOs including the one from CAMP4 Therapeutics in toxicology evaluation which is the last step before filing for an IND and several other programs in development. CDKL5 deficiency disorder is also preparing for next-generation gene therapies, and we saw a presentation from UCDavis with a viral-delivered epigenetic gene therapy that can open up the inactive X chromosome to specifically drive expression of the CDKL5 gene copy and that is also in late preclinical stage.
So the future is now for Dravet syndrome, and around the corner for several other DEEs. Based on the speed of progress in Dravet syndrome, I estimate that the AES conference in about three years will probably be the one with clinical results for disease-modifying therapies in multiple DEEs since we are on track for multiple trials to start in 2025 and 2026. But hey, maybe things go even faster now that we know (and investors know) that cures, or life-transforming improvement across all disease domains, are a real possibility.

2 - BEYOND SEIZURES IN DEEs
I mentioned before that Dravet syndrome is not alone, and that if we broaden our focus to include other neurodevelopmental disorders that are often less featured at AES (despite also having epilepsy) we see that 2024 has been an unprecedented year when it comes to showing neurodevelopmental improvements in monogenetic disorders with ASOs and gene therapies:
In Angelam syndrome, Ultragenyx and Ionis have announced that they are progressing to Phase 3 trials with their ASOs that increase UBE3A expression. Ionis showed data from their Phase 1/2 HALOS showing improvements in communication, cognition and motor function in Angelman patients ages 2-50. Ultragenyx also released data this year from their Phase 1/2 trial in Angelman patients ages 4-17, also showed rapid improvement across many disease domains.
Some diseases are not obvious targets for ASO treatments, and in Rett syndrome we have one of the first neurodevelopmental disorders to get clinical data from gene replacement gene therapies. Neurogene recently shared data from the first Rett syndrome patients (ages 4-7) treated with their viral gene therapy, also showing improvements in fine and gross motor skills, communication and cognition, while girls with Rett syndrome don’t re-gain skills after their early regression. Neurogene run into safety problems at a higher dose of the gene therapy, but the lower dose had substantial efficacy and should be a viable dose moving forward.
This brings us to 3 neurodevelopmental disorders that this year reached proof of disease-modification with genetically-targeted treatments, and their value goes beyond Angelman, Rett and Dravet syndrome because what they tell us is that the brain keeps much plasticity that is ready to go as soon as we get the right therapies.
To be able to document these improvements, companies had to move beyond seizure counting and figure out which scales could be used for Phase 3 trials in these disorders to document non-seizure improvements. It seems from Dravet syndrome that the non-seizure scale for the Phase 3 will be the Vineland-3 scale, while in Angelman both Ultragenyx and Ionis are choosing the Bayley scale for their Phase 3 trials. In fact all these companies were able to interpret their open-label Phase 1/2 trials because there had been previous efforts to document the performance of this type of scale in a particular type of Natural History Studies called endpoint-enabling studies, showing minimal or no skill development beyond certain age for these disorders. None of these findings would have been possible without those natural history studies.
In Dravet syndrome, the endpoint-enabling studies were run by Stoke and Encoded specifically targeted to the ages of their interventions. Encoded deep-phenotyped Dravet patients under 5 in their ENVISION study, and was able to show an inflection point around 2 to 2.5 years of age when patients slow down in their neurodevelopment despite the best standard of care. Stoke further extended this age range in their Butterfly endpoint-enabling study to show lack of changes in seizure frequency and skills over 12 months in this population, which leads to a growing neurodevelopmental gap when compared to the neurotypical population. These results were presented at AES as the reference to interpret the progresses that we are now seeing with the disease-modifying therapies and also to justify trial design.
While in Dravet syndrome those studies were initiated and run separately by two biotech companies, in CDKL5 deficiency disorder the Loulou Foundation was able to convene several companies in their space to participate jointly in a single endpoint-enabling study called the CANDID study by creating a pre-competitive consortium. This year the CANDID consortium presented the baseline results from this study at AES, which includes scales like the Vineland-3 and the Bayley that seem to be becoming the frontrunners for Phase 3 trials in this space, and the study is now collecting longitudinal data for all these scales.
And other communities chose to run before the companies in their space were ready, and to launch endpoint-enabling studies to start documenting the suitability of those preferred-scales for their particular disorder and their progress over time. This is the case of the STARR study for STXBP1-related disorders and the ProMMiS study for SYNGAP1 which were also presented at AES.
These studies are key for a disease to be “trial ready” for beyond-seizure trials, and key for the transition from treating only seizures to treating the syndrome.

3 - BEYOND THE LARGEST DEEs
At this point you are probably tired of hearing me mention the same group of DEEs over and over. That is because they have the most patients and the most advanced pipelines, but 2024 has been, again, a very different year when it comes to going beyond the top DEEs.
It started with Longboard pharmaceuticals announcing last summer that they have agreed with FDA to run a Phase 3 trial for bexicaserin (a second-generation fenfluramine) in “all DEEs” as opposed to cherry picking two or three of the largest syndromes. Longboard, recently acquired by Lundbeck, was one of the stars at AES and I have to praise them for making the time to meet with as many of the rare disease advocacy groups as they could during the conference, which is NOT an easy task when you have expanded your scope to include literally all DEEs!
The first-ever Phase 3 trial for “all DEEs”, called DEEp OCEAN (with bexicaserin), has already started in the US and will open up sites in Europe and beyond in this coming 2025. And at least one other company has already announced that they will be following on these steps: Praxis expects to being enrollment into the EMERALD trial with relutrigine (a next-generation sodium channel blocker) in the first half of 2025.
This means that for so many other DEEs, include very rare ones, the future of being chosen for clinical trials is also now, and arrived in 2024. What a year.
These combined “all DEE” trials are possible because they are counting seizures, and their approval will be for the treatment of seizures in DEEs. But we were just talking about how DEEs are “neurodevelopmental disorders with seizures” and we really need treatments that address the neurodevelopmental part.
I expect the next few years to get us closer to clinical trials across DEEs with treatments that can target some of the core biology of the diseases and that will need to show improvement in non-seizure outcomes. One example are genetic approaches like the one from Tevard Bio, that rescues non-sense (premature stop) mutations regardless of the gene that has them. Tevard was present at AES including at the SYNGAP1 conference. Another example are small-molecule drugs that might improve seizures and non-seizure outcomes like the KCC2 activators (some were presented at AES, other companies were in attendance but not presenting). For all those, we will need to identity clinical scales that work across different DEEs and to come up with a “core outcome set” for the non-seizure aspects of DEEs which might be a combination of some elements of the Vineland-3 and the Bayley, and perhaps other scales. Here again, we rely on those endpoint-enabling studies that the different communities are running to help us unlock the next-generation treatments for DEEs that can work across disorders and not only on their seizures.
But that is the future for the next few years. Today we celebrate a 2024 that was unprecedented in terms of tangible progress and change, with Stoke’s data adding to the news on Angelman and Rett syndrome that makes us believe in disease-modifying treatments, and with Longboard opening the door for innovation to reach everyone. What a year.
Ana Mingorance, PhD
Disclaimer: I write these texts with the parents of people with rare epilepsy syndromes in mind, so excuse also my lack of technical accuracy in parts.

MAIN LESSONS FROM THE 2024 CDKL5 FORUM
For the past ten years the Loulou Foundation hosts an annual meeting where scientists and drug developers working on CDKL5 deficiency, together with representatives from patient organizations, meet to discuss the latest advances.
Here are the main news and take-home messages from the 2024 CDKL5 Forum that took place in October 28-29 in Boston
For the past ten years, the Loulou Foundation has hosted an annual meeting, the CDKL5 Forum, where scientists and drug developers working on CDKL5 Deficiency Disorder (CDD), together with representatives from patient organizations, meet to discuss the latest developments in the field and to advance towards treatments and cures. You can find summaries from some of the last meetings here: 2018, 2019, 2020, 2021, 2022 and 2023.
The 2024 CDKL5 Forum edition took place October 28-29 in Boston, and will stay in Boston next year for the 2025 edition. This was the largest Forum to-date, with over 200 participants including representatives from 33 companies and 18 national patient groups. This was also a special edition, as this was the 10th CDKL5 Forum (with the first one taking place 9 years ago). I will try to summarize the main take-home messages from this year’s Forum. This won’t cover all the presentations, but rather focus on the main themes that we saw and the main progresses – which are always many.
1. New interesting learnings about CDKL5 biology and CDD

Image: Researcher Maria del Carmen Martin Carrascosa (CIPF), Forum Director Dan Lavery (from Loulou Foundation), and research doctor Josephine Thinwa (UTSouthwestern).
Sila Ultanir introduced a session on the biology of CDKL5 saying that a few years ago we only knew that CDKL5 was missing, so we could only think of putting back CDKL5 as a way to cure the disease. But today we know much more about what it does in cells, so this opens up different entry points to try to correct some aspects of the disease without needing to put back CDKL5 yet.
We learnt from Oguz Kanca from Baylor that there are patients with gain of function mutations in CDKL1 and CDKL2 that have neurodevelopmental syndromes so CDKL5 is not the only important gene in this family. And we learnt from Maria del Carmen Martin Carrascosa that fruit flies without CDKL5 have spontaneous epilepsy and repeated stereotypies so they seem to capture better the lack of CDKL5 that mice do. However in any animal that came in evolution before jaws appeared (yes, jaws), CDKL5 is acting almost like a CDKL1-4 which don’t have the full scope of functions of our CDKL5.
One of those potential redundant CDKL genes is CDKL2, so last year we considered if increasing CDKL2 could be a possible therapeutic approach when CDKL5 is missing. This year Kevin Dempster presented what happens if you get a mouse missing both CDKL5 and CDKL2… does it get even worse? The answer is yes but interestingly CDKL2 seems to only do SOME of the jobs that CDKL5 does in the cell, not all of them, notably not the job of changing activity of calcium channels and glutamate receptors. So in a way, increasing CDKL2 has the potential to rescue some of the consequences of missing CDKL5, but other consequences still need to be addressed separately, as is the case of still needing to inhibit Cav2.3 (more on this later).
On the biology side, we learnt from Josephine Thinwa that CDD patients might be more sensitive to viral infections, causing a larger inflammatory response, and from Lauren Orefice that lack of CDKL5 in different sensory neurons makes people with CDD more sensitive to textures, sensations (tactile sensitivity, like a tag in your clothes) and more prone to GI pain. All these seem to be sensory problems caused by CDKL5 lack in sensory neurons.
There were also different presentations during the Forum and pre-Forum meeting that point to the thalamus as an important brain part in CDD, as part of the thalamocortical connection. For example Rachel Oren presented that the cortical Visual Impartment is not in the eye (which is fine) or in the neurons that go from the eye to the thalamus, but somewhere later by the time the signal gets to the visual cortex. Another important brain component seems to be interneurons, shown by Tim Benke using a CDKL5 inhibitor in brain slices, which could help explain why some of the GABAergic drugs including ganaxolone are the best for CDD.
2. Beyond the research toolbox: the year of platforms
a. Cells and mice
One of the main goals of the Loulou Foundation early on was to make sure that any scientist anywhere in the world can work on CDD. This meant creating a collection of research tools like animal models and making them open-access. Can we see where CDKL5 is in this cell? Can we see if it is active? Can we see the consequences of missing CDD? And what does this mean about this organ physiology? we can’t answer this without cells and antibodies and mice.
This has led to a growing collection of animal models and cell models and antibodies which are available to the community. But this was perhaps the year of “platform as a service”, which is the next step once tools exist.

Image: slide from CDD mouse testing platform, slide from CDD iPSC-derived neuron platform, and researchers Liz Buttermore (BCH) and Jasmine Carter (Loulou Foundation) during the breakout session on iPS cells.
I want to highlight the iPSC-derived neurons from CDD patients which are running as a service at Boston Children’s Hospital Neuron Core led by Liz Buttermore, and the breakout session that Liz and Loulou Foundation’s Jasmine Carter led about the iPSC-derived models.
Liz walked us through all the crazy work that led to their very good in vitro epilepsy assay with patient-derived iPSC lines, and it was a great example of how hard is to standardize an assay of this type of have it be robust and reliable. Companies can now send their compounds to Boston to be run in this epilepsy-in-a-dish CDD model using microelectrode arrays.
We had a breakout session in the afternoon led by Liz and Jasmine to see how the academic and biotech community will want to work together in setting up these in vitro models. There were discussions about learning from each other successes and mistakes, and having a shared channel to ask questions since troubleshooting these models can get so tricky. The discussion was fantastic (and I’m sorry I missed the parallel breakout sessions).
Just like with cells, The Jackson Labs has set up a panel of mouse models of neurodevelopmental diseases that are now available for drug testing in a fee-per-service way. Rajat Puri presented data about their CDD mouse preclinical testing service, showing robust signal across mouse ages, mouse batches and even across lab researchers. Again, these standards are very tricky and it is powerful to have them available as a service as opposed to having to go through all the troubleshooting in each individual lab. The tests that I liked the most showed how bad CDD mice are at burying marbles and building nests, which are the mouse equivalent to activities of daily living for people.
And why are the services so important for companies working on treatments? Because having a mouse or a cell line available is like buying ingredients at the supermarket, we still need to cook them in the lab, but these in vitro patient models and complex behavioral mouse models are very elaborate and easy to get wrong, so having them running as a service is like offering restaurants where you can get the complex dish ready. It is much easier to go to a restaurant and order a paella than to spent two years learning how to cook a good one.
b. Patients
There is an observational clinical study in CDD designed to learn about clinical scales to measure symptoms other than seizures in trials, and to collect data that might serve as a control dataset for future clinical trials in CDD. It is called the CANDID study and I am very very proud of it, not only because I was part of the early team that saw the need for such study and got it started, but because it has evolved and it is now owned by the community. At the Forum, Xavier Liogier from the Loulou Foundation presented the results from the scales assessments in the CANDID study, which has more than 100 patients from the US, EU and MENA, and we learnt not only which scales could be used in a trial today for a gene therapy, but also the fact that 83% of patients would qualify for a trial if it asked for a minimum of 16 seizures per month. 83% IS A LOT! In other syndromes 25% or less of patients would have enough seizures for trials, and seizures are very likely to remain as the primary endpoint for trials, so in a way we have the perfect disease for clinical trials. This is important to know!

Image: Xavier Liogier (Loulou Foundation) presents an update on CANDID, and group discussion with doctors and patient representatives.
There was also a meeting for the CANDID study where Xavier and Maria Makarovskaya, the two professionals from the Loulou Foundation in charge of CANDID, asked clinicians in the study and representatives from the patient community about their experience in the study and their feedback. And this is where it became clear to me that CANDID was started by a small group of people, but it is now owned by a much much larger group, including the 112 patients that signed up and their parents, and all of the study investigators and their team of nurses and psychologists. This is huge, and a reason for many to be proud.
There is also a large fluid biomarker effort that was discussed in one of the breakout sessions. A fluid biomarker is a way to measure something in blood or CSF or other body fluid that shows that a therapy is working. For example sugar levels in blood serves as a biomarker for diabetes, while cholesterol in blood serves as a biomarker for cardiovascular risk. Maurizio Giustetto from Italy is looking at saliva to measure little vesicles that neurons spit out to talk to each other and that could potentially be picked in saliva (crazy!). And there is an international collaboration called ELPIS led by Massimiliano Bianchi that is collecting patient blood samples to check for potential biomarkers in plasma. They already found some potential plasma biomarkers, I wrote about that last year, and one year later the program is expanding to more countries and is considering a sample collection of blood and saliva from patients and siblings (as controls) during the Rome 2025 patient conference. Stay tuned for more information about that.
3. Therapies: the now, the near now and the blurry future
At the beginning of the therapies session, Omar Khwaja who is a doctor who has treated kids with neurodevelopmental disease and then became a drug developer, provided an overview of the CDD pipeline today.
We have ganaxolone approved in the US and Europe, and the CDD patients also use often Epidiolex off label, because it is approved for LGS and because there is good Phase 2 data for CDD. So we “almost” have two drugs approved. We then have clinical trials with fenfluramine (in Phase 3) and bexicaserin (about to start Phase 3 for DEEs including CDD). After that, there is a group of four “almost ready for trial” programs including three gene therapies.
In Omar words, we’ve come really far in these 9 years since the first Forum, with an entire landscape of programs in development including disease-modifying genetic therapies in late preclinical stage and multiple drugs in trials or approved. I invite you all to see the older summaries of the Forum to see this progress, including former trials that didn’t progress like ataluren (negative data in Phase 2) and soticlestat (positive data in Phase 2 but not chosen by the pharma for a Phase 3).
a. The now

Images: Sabine Bongardt from UCB Pharma, Randall Kaye from Longboard, and a slide from Elisa Borghi’s presentation on the pre-biotic and pro-biotic supplement trial.
UCB Pharma presented the progress with the fenfluramine Phase 3 trial in CDD, which is probably closing recruitment at the end of this year. They are particularly asking for children ages 1 to 2 to participate, with room for about 20 in the trial. This is very exceptional, because trials often only start at 2 years of age and in a disease like CDD that can start as early as in the first weeks of life, those two years can mean over one thousand seizures before the child could start taking fenfluramine (or any other drug with a minimum age of 2 years!). So if you know any family with a one year old with CDD and uncontrolled seizures please let them know about this trial (more info about locations in the trial website) so that this trial will complete recruitment at the end of the year.
And as one trial ends another one starts. Longboard Pharma is developing a drug called bexicaserin that is a second-generation fenfluramine, and it is starting a Phase 3 trial combining all epilepsy syndromes and in particular the large ones (including CDD) for patients ages 2-65 with at least 4 countable seizures a month. They are aiming for 80 trial hospitals, potentially over 100, and to start recruiting really soon. So check with your doctors if Longboard has reached out to them about this upcoming trial with bexicaserin called DEEp OCEAN.
By the way, Longboard has been quite popular recently because of choosing to combine all the epilepsy syndromes (all the DEEs) into a single trial and they even recently got bought by Lundbeck, the pharma company that makes clobazam. So there has been a lot of momentum around bexicasein. And Longboard received the 2024 CDKL5 Forum Company Making a Difference Award and CMO Randall Kaye talked about seeing an amber light as a sign to speed up, not to stop. Longboard is speeding up to treatments for all DEEs, even though the difficulty of the task ahead could have made others want to slow down (or turn away). For us in CDD, their Phase 3 means another chance to access a novel molecule in development before it gets approved.
And at the Forum we also had a very interesting presentation from Elisa Borghi about a clinical trial with a mix of pre-biotic and post-biotic supplements, so “food for your microbiome” and “products that good microbiome would release”. Their research was started by the Italian patient family group, and it is now in a Phase 2 trial with CDD patients to see how it might improve GI function and epilepsy, which seems to have a GI component. What I like of this program is that if the trial is positive, families can directly access the supplement and don’t need to wait for lengthy regulatory approval and price reimbursement negotiations as is the case for drugs. And there was another company in attendance although not presenting that is also targeting the microbiome, but using the good bacteria (so it is not a pre- or post-biotic but a biological treatment!). I hope that next year we also have them present and learn more about clinical interventions in this gut-brain area.
b. The near now
I don’t know if you have realized that we have now three gene therapies and one small molecule program all past clinical candidate nomination. “Clinical candidate” is how we call a drug or therapy that is already done with optimization and tweaking, and that scientists then transition to safety and toxicology testing in animals other than mice so prepare to file and IND (a permission to start trials).

Images top: Ultragenyx’s Emil Kakkis and slide from their gene therapy program. Slide from Lario Therapeutics.
Images bottom: UCDavis’ gene therapy slide and Kyle Fink. Slide from the Loulou Foundation and Penn gene therapy presentation.
One of these is the gene therapy for Ultragenyx, which CEO Emil Kakkis explains that already reaches enough number of neurons in pigs (and is safe) to look ready for trials, and in a few months they hope to make a decision about potential methods to improve the brain distribution of the virus prior to then progressing to an IND. He also showed us great results that Ultragenyx is having in other three neurological diseases like Angelman syndrome where Ultragenyx is progressing to a Phase 3, and left us with the hopeful learning that “the brain is more plastic and more capable than we think”.
A second program in this near now category is the gene therapy from the Loulou Foundation, funded via its subsidiary Elaaj and created by the Gene Therapy Program at UPenn under the direction of gene therapy super-expert Jim Wilson. Jim recently left Penn to start a company called Gemma Therapeutics so the presentation of this gene therapy was done by Janine Lamonica, the leading scientist for this program at Gemma. This gene therapy looks like a solid clinical candidate that is ready to take forward. Both the Ultragenyx and the Loulou Foundation gene therapies use a copy of human CDKL5 gene inside of an adeno-associated virus (AAV) and are administered via injection into the cisterna magna, the big pool of CSF that you can access at the nape, in between your neck and your skull (via ICM). So these gene therapies are fairly similar.
A third gene therapy that is now moving into toxicology is the X-reactivation CRISPR 2.0 approach from Kyle Fink at UCDavis that uses a modified CRISPR to find the second CDKL5 gene in cells, which is inactivated as part of the inactive X chromosome, and makes the cell read it. Instead of find-and-cut this is a find-and-activate type of CRISPR, and to get it to neurons the scientists are using two AAV virus, each carrying half of the CRISPR sequence (actually millions of virus of each, not only one of each, so that when they infect the same neuron they can make the find-and-activate type of CRISPR). Their mouse rescue data is beautiful and they are seeing success in adult female mice, again showing that it is probably never too late for CDD!
And the small molecule program that has already nominated a candidate (a final molecule) is the Cav2.3 inhibitor program from Lario Therapeutics. Two years ago we learnt that Cav2.3 is a channel that makes glutamate neurons fire and that is usually controlled by CDKL5 as a break, bringing down its activity. In the absence of CDKL5, Cav2.3 is too active and excitatory neurons go crazy, so we would need inhibitors. Last year we saw some early data with those inhibitors made by Lario, and this year they showed us a beautiful dose-response activity in glutamatergic neurons made from CDD patient cell lines, so the more drug the more correction of crazy excitation. This is a drug, not a gene therapy, which makes it very attractive to combine with other treatments.
c. The blurry future
Here is where we have all the other therapies including gene editing (the one that fixes your DNA sequence, by David Liu), or the gene therapy that fixes non-sense mutations (by Tevard), or the early-stage ASO approaches that are being attempted to rescue CDKL5 or increase CDKL2, and even the efforts to make CDKL5 protein in the lab to then give it to the brain (enzyme replacement therapy). There are also talks about changing the virus type to the ones that can be administered via blood injection and then cross to the brain, so that we don’t need to inject gene therapies directly into the brain. Many people are working hard in all those avenues, but we don’t know IF any of them will eventually get to trials or WHEN they might get to trials. So what we know is that today, the best gene therapy type that medicine knows how to do is to put a gene copy inside of an AAV9 virus or similar and to administer this gene therapy directly into the brain, like the three CDD gene therapies in the “near now” category. Anything else, is still a blurry (but hopeful) future.
4. What’s good enough and the big head problem
One thing that became clear in the discussions with the speakers and the industry panel is that because AAV gene therapies can only be done once, we really need to be sure that we got the right dose and distribution before trials. In other diseases where the main “gene therapy” type is an Antisense Oligonucleotide (ASO), which is like a piece of DNA that you inject into the spine once every few months, you can go to trials earlier and figure out the right dose there, but you cannot do this in viral-delivered therapies which are once-and-done. I will give you two real examples for this go-to-trials speed advantage with ASOs: (1) in Angelman, Ultragenyx went into the clinic initially with a dose of ASO too high, they saw some localized inflammation in the spine and scary leg paralysis but that was transient, and they could continue dosing those same patients with lower doses. (2) In Dravet syndrome, Stoke Therapeutics started trials thinking that the good dose would be 30mg, it wasn’t, so they went up to 45mg to try to see efficacy and still wasn’t enough, so then they started using 70mg as the dose and finally got impressive efficacy. In these two cases, the first dose was “wrong”, but it wasn’t a big problem because you keep dosing ASOs every few months so you can always go up or go down. But in CDD we haven’t had good shots on goal until recently to use ASOs, and we are instead using viral-delivered gene therapies as our first gene therapies, and because in those you literally only get one shot we are taking much longer to start trials because they have to be right from the start.
So how do we know when it is good enough? In the case of CDD the main challenge seems to be being confident that we are reaching enough neurons, because mice have small brains that are easy to cover with the virus but we have much much larger brains (a 5 year old has a brain 2,500-times larger than a mouse brain). So that is our challenge, to have so much virus spread throughout the brain in pigs and monkeys (mid-size brain) that we can trust we can jump to humans and get enough brain infection to see good efficacy.
There was quite a bit of debate about “what is good enough” to take those gene therapies to trials, which the consensus answer being that we need to be “comfortably sure” before starting trials. It still looks like it will come in the near future, but this is why it is so hard to pin-point exactly when. And the feedback from the industry experts in the panel was that even though we know the future will bring better gene therapies than the current most-advanced ones, this should not prevents us from starting trials with the almost-ready ones as soon as they are ready. Because patients don’t have so much time to wait in particular if they are young, and because we don’t know what future technologies will bring us and when.
As I write this, I am also thinking that we need to work hard on getting non-viral therapies to move to trials because they can be combined with the viral gene therapies, while scientists have not yet found a way to be able to give more than one viral gene therapy to the same person (the “redosing” problem, see also the next section). There are projects that were not presented at the Forum because they are early stage but they fit this need very well, such as the development of ASOs to increase CDKL2, ASOs to fix CDKL5, and making the CDKL5 enzyme in the lab to then infuse it into the brain (enzyme replacement therapy). I hope we can review these next year.
5. How the patient community can help

Industry panel, from left to right: Ralf Schmid (Novartis), Sharyl Fyffe-Maricich (Ultragenyx), Randall Kaye (Longboard), Brian Moseley (UCB) and moderator Majid Jafar (Loulou Foundation)
We had a panel with industry experts in drug and gene therapy development to see how we, the community, can help get new treatments forward faster. Some messages were:
EDUCATE/RESEARCH: Help families understand that being part of research should be part of the treatment paradigm. It doesn’t need to be get into clinical trials with drugs, but perhaps observational clinical studies like CANDID, or biomarker studies like ELPIS. If patients don’t participate then we can’t get that next generation of treatments.
RESEARCH: Help validate scales and outcome measures that can go beyond seizures, so that the new therapies can show that they are better than the current seizure therapies (I think we are already doing this well with the U01 and the CANDID studies and with ELPIS)
EDUCATE families about gene therapies: what can we realistically expect, what does the journey of a trial for a gene therapy looks like…
ADVOCACY: Some challenges that we can’t fix but can perhaps influence are getting regulators to understand what we need for gene therapy clinical trials, and also the problem of pricing for gene therapies.
RESEARCH: also beyond our reach but where we can join researchers and other communities, is the problem of figuring out a way to make gene therapies redosable. If we know that we have a second-shot, and it is not only a one-shot goal, it will make it easier for people to participate in trials and for companies to run those trials. One way that we could help this is for example to have a focused workshop or session at the next Forum about the immune side of gene therapy and how to be able to one day re-dose patients.
I will add two things that they didn’t say but that I think are important roles that the community also plays towards getting new treatments:
RESEARCH/ COMMUNITY: I noticed that several of the presentations were on research projects that had initially been seeded by some of the national patient groups, like Canada, Italy and Spain. So this goes to show that seeding research is another important role of the patient community that can start the building of a mountain with the first grain of sand. I think this is a very important function of patient groups also, to seed CDD research in their countries.
COMMUNITY: We really need to find more patients. It is clear that there are more patients living with CDD that don’t know their diagnoses yet, than those who already have the correct diagnosis. An undiagnosed patient is a person who might be in the wrong treatment, who will miss all these trials, and who can’t help us solve the science questions (including the accurate epidemiology of the disease). I believe this needs to be addressed at a country level so I am adding it to our to-do lists!
6. The patient community: belonging, believing, and getting work done
The Forum traditionally opens and closes with the voice of the patient, and this year opened with Lily Howard telling us her experience as a sister to Harper, a little girl with CDD. Lily spoke about isolation and forced maturity, and feeling different from other kids and not able to connect. That’s why she has created an event for siblings, so that they get to create a sense of belonging.

Image: Majid and Lynn Jafar (Loulou Foundation, parents to Loulou); Lily Howard (Hope-4-Harper, sister to Harper); Andreas and Ana Born (CURE5; parents to Siena); and Heike Knip (CDKL5 Alliance President, dad to Valentina)
During the big gala dinner night, the patient award went to Ana Carolina and Andreas Borg, the parents of little Siena, who talked about the initial shock of the diagnosis, discovering a world that they didn’t know existed, to then finding purpose and a sense of agency in advocacy. I will share one beautiful sentence from Andreas: “all solvable problems can be solved if enough talented people put their energy into them”, and one from Ana: “we always look for heroes, and I’m proud I’m raising mine”. That’s the magic night of the Forum where we all end up crying and at the same time believing in a wonderful future that we can build.
To close the Forum, the outgoing Chair of the CDKL5 Alliance, Heike, spoke once again about the need for Unity in the patient community and shared the highlights from the last year work by each patient group in the Alliance. He also reminded us that we should try to improve quality of life, not just focus on gene therapies, with the example for how his daughter Valentina has gone from having 3-4 seizures a day to having 3-4 seizures a week in one clinical trial, so every bit that we can do helps a lot. This call echoes the words from Lily at the opening of the Forum, when she told us that even assistive technologies (not even medicines) can improve quality of lives for patients and their families. It is a beautiful reminder that at the end of the day, we are treating a person, not just a gene.
Last, Majid Jafar, co-Founder of the Loulou Foundation, wrapped up the Forum asking us all to meet at the CDKL5 Alliance meeting in Rome in June 2025, and announcing that the next CDKL5 Forum will return to Boston in October 27-28 of 2025. And he left us with a call to action: “we’ve come so far… and there is still so much to do. Let’s get it done”.
So we get back to work with our heads full of ideas and our hearts full of friends. I look forward to seeing you all in Rome and in Boston next year.
Ana Mingorance, PhD
Disclaimer: This is my own summary and key learnings, and not an official text about the Forum by the Loulou Foundation. I write these texts with the parents of people with CDD in mind, so excuse also my lack of technical accuracy in parts.

REPASO DEL FORO CDKL5 2024
La décima edición del Foro CDKL5 tuvo lugar en Boston, los días 28 y 29 de octubre de 2024. El Foro es una reunión anual que organiza la Fundación Loulou y en la que científicos y miembros de la industria farmacéutica se reúnen con representantes de la comunidad de pacientes para repasar los últimos avances en el campo.
Este es un repaso para los grupos de pacientes de las principales novedades del Foro CDKL5 2024.
Hace ya diez años que la Fundación Loulou organiza una reunión anual, el Foro CDKL5, donde los científicos de academia y de industria trabajando en el síndrome de deficiencia en CDKL5 (CDD), junto con representantes de los grupos de pacientes, se reúnen para compartir las últimas novedades y avanzar hacia tratamientos y una cura. Tenéis el resumen de los años pasados aquí: 2018, 2019, 2020, 2021, 2022 and 2023.
La edición de 2024 tuvo lugar en Boston los días 28 y 29 de octubre, y volverá a Boston otra vez en 2025. Este ha sido el Foro más numeroso, con más de 200 participantes incluyendo representantes de 33 empresas y 18 grupos de pacientes. Esta fue también una edición especial porque marca el décimo Foro (hace 9 años del primer Foro). Así que voy a intentar resumir las conclusiones principales de la edición de este año, no incluyendo todas las presentaciones sino centrándome en los temas principales y los progresos que vimos, que son muchos.
1. Seguimos aprendiendo sobre CDKL5 y sobre el síndrome por deficiencia

Imágenes: la investigadora Maria del Carmen Martin Carrascosa (CIPF), el Director de Foro Dan Lavery (from Loulou Foundation), y la medico e investigadora Josephine Thinwa (UTSouthwestern).
Sila Ultanir abrió una sesión dedicada a la biología de CDKL5 diciendo que hace unos años solo sabíamos que faltaba CDKL5, así que para tratar la enfermedad solo podríamos pensar en poner de vuelta ese CDKL5 pero ahora sabemos mucho más sobre lo que hace en las células, lo cual nos abre oportunidades para intentar corregir esas alteraciones directamente sin necesidad de devolver CDKL5, y nos da muchas más opciones.
Aprendimos de Oguz Kanca de Baylor que hay pacientes con mutaciones de ganancia de función en los genes CDKL1 y CDKL2 que también tienen síndromes de neurodesarrollo, con lo que CDKL5 no es el único gen importante de la familia. Y aprendimos de Maria del Carmen Martin Carrascosa del centro Pincipe Felipe en Valencia que las moscas de la fruta sin CDKL5 tienen epilepsia y estereotipias, con lo que parecen mejor modelo de CDD que los ratones. Y sin embargo en todos los animales que aparecieron en la evolución antes de que aparecieran las mandíbulas (si, las mandíbulas) usan CDKL5 casi como si fuera un CDKL1-4, que no hacen tantas funciones como nuestro CDKL5.
Uno de esos posibles genes CDKL redundantes es CDKL2, que ya el año pasado hablamos de que quizás podría ser una diana a modificar con terapias cuando nos falta CDKL5. Este año Kevin Dempster presentó lo que ocurre si a un ratón le quitas el CDKL5 y también el CDKL2. ¿Está peor aún ese ratón? La respuesta es que si, está peor, pero curiosamente CDKL2 solo hace algunas de las cosas que hace CDKL5, le faltan algunas funciones importantes como regular el canal de sodio Cav2.3 o los receptores de glutamato. Así que de cierta forma aumentar CDKL2 podría suplir alguna (pero no todas) de las funciones de CDKL5, pero aún tendríamos que corregir algunos elementos por separado por ejemplo aún necesitando inhibidores de Cav2.3 (os cuento más de esto más adelante en el texto).
Sobre la biología de la enfermedad, aprendimos de la experta en inmunología Josephine Thinwa que las personas con CDD podrían ser más sensibles a las infecciones víricas, que resultarían en una reacción inflamatoria más grande de lo normal, y de Lauren Orefice aprendimos que la falta de CDKL5 en las neuronas sensoriales es la responsable de que las personas con CDD tiendan a tener aversiones a ciertas texturas y roces (por tener la sensibilidad táctil alterada) y más propensos al dolor intestinal.
Otras presentaciones apuntaron a partes del cerebro importantes para el síndrome, como el tálamo, que Rachel Oren estudió como parte de la ceguera cortical (la señal del ojo llega bien hasta el tálamo y es en la conexión tálamo-cortical donde falla), o las neuronas GABAérgicas (las inhibitorias) que Tim Benke vio que eran responsables de la hiperexcitabilidad neuronal. Quizás por eso los fármacos que refuerzan la actividad de GABA como ganaxolona sean de lo mejor que hay para CDD.
2. Más que herramientas: el año de las plataformas
a. Células y ratones
Uno de los principales objetivos de la Fundación Loulou desde el principio es asegurarse de que cualquier científico en cualquier parte del mundo pueda investigar en CDD. Y eso ha implicado generar una colección de herramientas de investigación como anticuerpos para ver proteínas, y células pluripotentes de pacientes para convertirlas en neuronas en cultivo, y ratones con CDD. Y sobre todo hacer que sean de acceso abierto todas herramientas.
Y esto nos ha llevado a una colección importante de anticuerpos y células y ratones que están a disposición de la comunidad científica. Pero este año va un paso más allá y ha sido el año de las plataformas como servicio.

Imagen: diapo de la plataforma de evaluación de terapias en ratones con CDD; diapo de la plataforma de evaluación de fármacos en células derivadas de pacientes; y las investigadoras Liz Buttermore (BCH) y Jasmine Carter (Loulou Foundation) durante la sesión paralela sobre células iPS.
Quiero destacar las células iPS a partir de pacientes que se están usando como servicio en el Boston Children´s Hospital por Liz Buttermore, y la sesión de trabajo paralela que Liz y Jasmne Carter de la Fundación Loulou facilitaron durante el foro. Por cierto las células iPS son células pluripotentes que se hacen desprogramando células de la sangre o piel de personas (en este caso de pacientes) para que se puedan convertir en lo que queramos en laboratorio, y en este caso queremos neuronas, claro.
En su charla, Liz nos explicó el trabajo de locos que ha sido conseguir hacer un modelo de epilepsia en placa de Petri usando las neuronas de pacientes porque estandarizar el protocolo de cultivo y de diferenciación en neuronas y su medición ha sido una odisea, pero al final es muy robusto y reproducible. Las empresas que están testando terapias para CDD ahora ya las pueden mandar a Boston para probarlas en este modelo de epilepsia en placa de Petri donde miden su actividad usando microelectrodos.
Y por la tarde tuvimos la sesión de trabajo con Liz y Jasmine Carter para ver como la comunidad académica y las empresas pueden trabajar de manera conjunta en desarrollar y utilizar estos modelos in vitro. Hubo debate sobre cómo aprender de los logros y errores de los demás, y crear algún canal de comunicación de grupo para poder hacerse preguntas porque es tan complicado trabajar con estas células. Me pareció una sesión fantástica y lo siento por las que ocurrieron en paralelo y que me tuve que perder.
Y similar a lo que han hecho con estas células, los Laboratorios Jackson han creado un panel de diferentes modelos de ratón de enfermedades de neurodesarrollo que está también disponible para probar fármacos bajo un modelo de pago por servicio. Rajat Puri nos presentó datos de plataforma preclínica de prueba de eficacia en ratones con CDD donde consiguen unos fenotipos muy robustos en ratones de edades diferentes, lotes diferentes, e incluso testados por técnicos de laboratorio diferentes. De nuevo estos estándares suelen ser muy complicados y de ahí el valor de tenerlo disponible como servicio de pago en vez de tener que pasar por la odisea de adaptar el modelo en cada laboratorio. Los tests que más me gustaron son los que enseñan que los ratones con CDD son malísimos a la hora de enterrar bolitas en el serrín de su caja o de hacer nidos con papel para dormir, que serían el equivalente ratón a las actividades diarias que hacemos las personas en nuestro día a día.
¿y por qué es tan importante tener un servicio? Porque tener un ratón o una línea celular es como tener un ingrediente comprado en el supermercado, te lo tienes que llevar a tu laboratorio y cocinarlo, pero en el caso de estos servicios con células o ratones para medir eficacia de terapias son tan tan complicados que es fácil usarlos mal, así que tenerlos disponibles como servicios es como tener un restaurante donde puedes tener el plato ya hecho por muy complejo que sea. Y es más fácil ir a un restaurante a comer una paella que pasarse dos años aprendiendo a cocinar una decentemente.
b. Pacientes
Hay un estudio observacional en CDD llamado CANDID que mide la capacidad de diferentes escalas motoras, cognitivas y conductuales para capturar estos aspectos de CDD en pacientes, y para recoger datos que puedan en un futuro servir de grupo control en ensayos clínicos. Y es un estudio del que estoy muy orgullosa no solo porque fui parte del equipo original que lo ideó y diseñó sino porque se ha convertido en un estudio que la comunidad de pacientes ha adoptado. En el Foro, Xavier Liogier de la Fundación Loulou nos presentó los resultados con las escalas que ya han sido probadas en más de 100 pacientes en EEUU, Europa y oriente medio, y como no solo ya sabemos que escalas usar para ensayos, sino que resulta que hemos visto que si los ensayos clínicos siguen pidiendo un mínimo de 16 crisis epilépticas al mes, el 83% de los pacientes cualificarían para el ensayo. ¡¡83% es un porcentaje enorme!! En otros muchos síndromes suele ser menos de un 25% que cualifica, y como los ensayos clínicos suelen usar la epilepsia como su medida de eficacia principal eso nos hace un síndrome ideal para ensayos clínicos. ¡Esto es importante que se sepa!

Imagen: Xavier Liogier (Fundación Loulou) presenta los resultados hasta ahora de CANDID, y un grupo de médicos y representantes de pacientes participan en una discusión sobre el estudio.
También hubo una reunión sobre el estudio CANDID donde Xavier y María Makarovskaya, los dos profesionales dela Fundación Loulou a cargo del estudio, preguntaron a los médicos y a los representantes de pacientes sobre su experiencia con el estudio hasta ahora y su input sobre posibles ajustes y cambios. Y es en ese momento cuando me di cuenta de que el estudio CANDID lo empezamos un grupo pequeño de personas pero ahora lo cuida un grupo muchísimo más grande de gente que incluye los 112 pacientes y sus familias y sus médicos y los neuropsicólogos del estudio y enfermeras… todos super involucrados en el éxito del estudio. Es una pasada, y una razón para que muchos estemos orgullosos.
También hay varios proyectos en torno a la obtención de biomarcadores en fluidos que se trató en otro de los grupos de trabajo paralelo. Un biomarcador en fluido es algo que se puede medir en sangre o líquido cefalorraquídeo o cualquier otro fluido corporal y que nos pueda indicar si una terapia está funcionando. Por ejemplo mirando glucosa en sangre tenemos un biomarcador de diabetes, y mirando colesterol sabemos cómo estás de salud cardiovascular. El italiano Maurizio Giustetto está mirando muestras de saliva para ver pequeñas vesículas que las neuronas liberan para hablarse las unas a las otra y que potencialmente podrían llegar hasta la saliva (esto es flipante). Y Massimiliano Bianchi lidera el estudio ELPIS que es una colaboración internacional donde miran en plasma (en sangre) cambios que indiquen lo que pasa en el cerebro. Ya el año pasado nos contaron cambios que se están viendo en sangre, y que hay con confirmar con una cohorte más grande de pacientes, y este año están expandiendo el estudio para incorporar más países y hospitales. También existe la posibilidad de recolectar muestras de sangre de pacientes y sus hermanos en el congreso de la Alianza CDKL5 del año que viene en Roma, así que estar atentos a más noticias sobre este proyecto.
3. Terapias: las de ahora, las de casi ahora y el futuro borroso
Al principio de la sesión sobre terapias, Omar Khwaja que es un médico que cuidaba de pacientes con enfermedades de desarrollo y luego se ha convertido en desarrollador de terapias, nos dio un resumen de las terapias en desarrollo para CDD.
Ahora mismo tenemos ganaxolona, aprobada en EEUU y en Europa aunque aun no disponible en Europa comercialmente, y muchas de las familias con CDD están tomando Epidiolex ya sea como Lennox-Gastaut ya sea como fuera de prescripción. Con lo que “casi” tenemos dos fármacos aprobados. Luego tenemos el ensayo clínico de fenfluramina que está terminando, el de bexicaserina a punto de empezar (para síndromes con epilepsia incluido CDD). Y tras esos tenemos un grupo de 4 terapias “casi listas” para ensayos, incluidas tres terapis génicas.
En palabras de Omar, hemos avanzado muchísimo en estos 9 años desde el primer Foro, con una colección de terapias en desarrollo incluidas terapias génicas en las últimas fases pre-clínicas y varios fármacos en ensayos o aprobados. Os animo a leer las entradas del Foro de años pasados para ver este progreso, que incluye programas que no avanzaron como ataluren que falló en Fase 2 y soticlestato que salió positivo en Fase 2 pero la empresa eligió no seguir hacia Fase 3.
a. Las de ahora

Imágenes: Sabine Bongardt de UCB Pharma, Randall Kaye de Longboard, y una diapo de la presentación de Elisa Borghi sobre el ensayo con el suplemento pre-biótico y post-biótico.
UCB Pharma presentó el progreso con el ensayo de Fase 3 de fenfluramina, que posiblemente cierre reclutamiento al final de este año. Hicieron un llamamiento en especial para niños de entre 1 y 2 años de edad de los que esperan poder incluir una veintena en el ensayo. Esto es muy excepcional, la mayoría d ellos ensayos empiezan a partir de los 2 años, y en una enfermedad como CDD que debuta incluso en el primer mes de vida, esperar a los dos años puede implicar más de mil crisis epilépticas hasta poder tomar fenfluramina (o cualquier otro fármaco que generalmente solo está aprobado para usar a partir d ellos dos años). Así que si conocéis alguna familia con un niño o niña con CDD de esa edad decidles de este ensayo clínico para que puedan cerrar reclutamiento a final de año con suficientes casos de menos de dos años y permitir una aprobación desde esa edad.
Y mientras acabamos este ensayo empezamos otro. La empresa Longboard está desarrollando un fármaco llamado bexicaserina que es una fenfluramina de segunda generación, y que está a punto de empezar un ensayo de Fase 2 donde juntan todos los síndromes con epilepsia, con especial interés en los más comunes como CDD. Buscan pacientes de 2 a 65 años, con cuatro crisis contables al mes como mínimo, y planean abrir el ensayo en 80 hospitales, quizás más de 100, y empezar pronto. Así que preguntad a vuestro médico si están en contacto con Longboard para este ensayo clínico con bexicaserina llamado DEEp OCEAN.
Por cierto Lonboard ha ganado mucha popularidad recientemente, primero por la decisión de ir a por todos los síndromes juntos, y luego porque han sido comprados pro la empresa farmacéutica Lundbeck que es la que hace clobazam. Longboard recibió el premio Foro CDKL5 de 2024 a la “Empresa haciendo una diferencia” y su director médico Randall Kaye habló de interpretar un semáforo en ambar como una señal para acelerar (que dice que es lo que hace la gente en ciertas ciudades). Explicó que la idea de convencer a los reguladores de dejarles juntar todos los síndromes en un ensayo para que así el fármaco se apruebe para todos es algo complejo y amucha gente les daría que pensar y frenarían, como con un semáforo en ambar, pero que Longboard decidió ir a por el desafío y acelerar la posible aprobación de la primera terapia para todos los síndromes. Para nosotros este ensayo es otra oportunidad de acceder a un fármaco nuevo años antes de que esté disponible comercialmente.
Y en el Foro tuvimos también una presentación muy interesante de Elisa Borghi sobre un ensayo que está empezando en Italia con un suplemente que mezcla pre-bióticos y post-bióticos, o sea “comida para las bacterias buenas” y “productos que liberan las bacterias buenas”. Este proyecto comenzó con financiación del grupo italiano de pacientes y ahora está en ensayo de Fase 2 para ver como mejoran la función intestinal y a epilepsia de los pacientes, que parecen estar relacionadas. Una cosa que me gusta de este estudio clínico es que si resulta positivo las familias pueden adquirir directamente el suplemento ya que no hace falta esperar a aprobaciones regulatorias y de precio que suelen tardar años en el caso de fármacos. Y teníamos otra compañía en el Foro, pero que vino sin presentar, que también está desarrollando una terapia dirigida a la microbiota intestinal pero usando bacterias de las buenas (o sea no es pre- ni post-biótico sino un tratamiento biológico). Yo espero que el año que viene si presenten y poder tener una sesión sobre las intervenciones clínicas para estudiar y tratar la interacción microbiota-cerebro.
b. Las de casi ahora
No se si sois conscientes de que ya tenemos cuatro programas incluidas tes terapias génicas pasadas la “nominación de candidato clínico”. Un candidato clínico es como llamamos en la industria a un fármaco o terapia que ya hemos terminado de diseñar y de optimizar y ya es final, con lo que lo pasamos a la etapa de estudio de toxidad en animales mayores que ratones para poder entonces pedir el permiso de usar en ensayos clínicos (la “IND” es ese permiso).

Imágenes arriba: Emil Kakkis de Ultragenyx y una diapo de su terapia génica. Diapo de Lario Therapeutics.
Imágenes abajo: Diapo de la terpia de UCDavis y Kyle Fink de UCDavis; diapo sobre la terpia génica de la Fundación Loulou y Penn.
Uno de esos es la terapia génica de Ultragenyx, que su CEO Emil Kakkis nos explicó que ya llega a suficientes neuronas en cerdos y que es segura, y esperan en unos meses decidir sobre posibles ajustes en la forma de administrarlo para ayudar a que llegue a más células antes de avanzar hacia ensayos. Emil también compartió los resultados que están obteniendo en su empresa en ensayos clínicos con otras tres enfermedades neurológicas y nos dejaba con la lección esperanzadora de que “el cerebro es más plástico y más hábil de lo que creemos”.
Un segundo programa en esta categoría de proyectos “casi ahora” es la terapia génica de la Fundación Loulou que desarrolla via su empresa subsidiaria Elaaj y que se está desarrollando en el Programa de Terapia Génica de UPenn bajo la dirección del super experto de terapias génicas Jim Wilson. Jim hace poco que ha dejado la universidad y se ha llevado el proyecto a una nueva empresa llamada Gemma Therapeutics, con lo que científico Janine Lamonica de Gemma presentó este proyecto. Esta terapia génica también tiene todo lo de toxicidad completo y con buenos resultados y es un candidato clínico que también está listo para avanzar hacia ensayos, aunque no dieron fechas.
Tanto la de Ultragenyx como la de Loulou son terapias génicas muy similares, consistentes en el gen human CDKL5 metido dentro de un virus adeno-asociado (AAV) y administradas mediante inyección directa en la cisterna magna que es el espacio lleno de líquido cefalorraquídeo que hay en la nuca.
Una tercera terapia génica que está ahora avanzando a experimentos de toxicología es la de reactivación del gen CDKL5 inactivo de Kyle Fink en UCDavis que usa una especie de CRISPR 2.0 para encontrar-y-activar en vez de encontrar-y-cortar que hacía el CRISPR original. Esta terapia va también dentro de virus AAV, en este caso dentro de dos porque el CRISPR 2.0 es muy grande (pero en una inyección hay millones de virus de cada uno de los dos tipos con lo que se juntan en las células). La terapia hace muy bien lo de encontrar el segundo CDKL5 que está inactivo en cada neuronas (solo leemos uno) y hacer que se lea de modo que todas las neuronas lean los dos, asegurando que todas las neuronas lean el bueno. Y sus experimentos en ratones son en ratones adultos donde funciona muy bien con lo que nos confirma que al menos en ratones nunca es demasiado tarde para restaurar la expresión de CDKL5 y ver mejoras clínicas (o preclínicas).
Y el cuarto proyecto que ha nominado un candidato clínico y que es un fármaco (no una terapia génica) es el de inhibidores de Cav2.3 de la empresa Lario. Hace dos años nos contaron que el canal de calcio Cav2.3 hace que disparen las neuronas glutamatérgicas (excitatorias) y que en general es controlado y frenado por CDKL5. Pero al faltar CDKL5 ese canal está demasiado activo y las neuronas excitatorias están descontroladas, por eso necesitamos inhibidores del canal. Hace un año nos dijeron que la empresa Lario estaba trabajando en inhibidores, y este año nos han enseñado una dosis-respuesta preciosa en neuronas de pacientes en la plataforma de epilepsia en placa de Petri de Boston. Dosis-respuesta significa que a más dosis más eficacia de corrección de la actividad descontrolada de esas neuronas de pacientes. Como esto es un fármaco y no una terapia génica tiene el atractivo de que se puede combinar más fácilmente con otros tratamientos.
c. El futuro borroso
En esta categoría tenemos todas las otras terapias en desarrollo, incluida la edición genética (que arregla la letras que está mal, que está haciendo David Liu), o la terapia génica que arregla las mutaciones non-sense (de la empresa Tevard), o los proyectos en fase temprana de ASOs para aumentar CDKL2 o arreglar CDKL5, y también los de terapia de reemplazo enzimático que es hacer la proteína CDKL5 en el laboratorio y dar infusiones periódicas a los afectados. También hay programas que estamos considerando con terapia génica pero vía intravenosa, usando virus que pasan de sangre a cerebro. Todas estas avenidas se están explorando pero no sabemos ni SI llegarán ni CUANDO llegarán a ensayos clínicos, por eso lo del futuro borroso. Lo que sabemos es que hoy por hoy, lo que la medicina sabe hacer bien es poner una copia del gen dentro del virus AAV9 o parecidos y administrarlas directas al cerebro, como hacen las tres terapias que están cerca de ensayos clínicos. Todo lo demás es aún un futuro borroso aunque esperanzador.
4. A qué llamamos “suficientemente bueno” y el problema de la cabeza grande
Una cosa que quedó claro con los ponentes y el panel de expertos de industria es que como estas terapias que usan virus AAV solo se pueden dar una vez, hay que estar super seguro de que sabemos la mejor dosis y vía de administración antes de decidir empezar un ensayo. En otras enfermedades donde la terapia dirigida a la causa usa otra modalidad, por ejemplo los oligonucleótidos antisentido (ASOs) que son como cachitos de ADN que se dan por punción lumbar cada par de meses, se puede ir a ensayos más rápido porque se puede ajustar la dosis en la clínica, pero eso no se puede hacer con los virus porque solo se pueden usar una vez. Os voy a dar dos ejemplos de esa ventaja de los ASOs a la hora de ir a ensayos más rápido: (1) en el síndrome de Ängelman, Ultragenyx empezó ensayos con un ASO con una dosis que resultó ser muy alta lo que produjo una reacción inflamatoria en la médula espinal que resultó en parálisis temporal de piernas, pero pudieron continuar el ensayo incluso en esos mismos pacientes dando una dosis más baja. (2) en el síndrome de Dravet, la empresa Stoke pensó que la dosis de su ASO buena sería la de 30mg, y empezaron con esa pero no vieron mucha mejora, así que subieron a 45mg y vieron un poco más pero no suficiente, y por fin con la dosis de 70mg dieron en el clavo con una eficacia muy alta. En ambos casos, la primera dosis la eligieron “mal”, pero no fuer gran problema porque como el ASO se da cada par de meses pudieron subir o bajar la dosis como fuera necesario. Pero en CDD no hemos tenido hasta ahora una buena oportunidad de usar ASOs, y los proyectos dirigidos a la causa más avanzados que hay usan todos virus, y esos solo se pueden usar una vez, así que con una oportunidad única tenemos que estar super seguros de que la dosis es la correcta, y por eso estamos tardando más que otros síndromes en empezar ensayos.
¿Y como sabemos cuando una terapia es “suficientemente buena”? en el caso de CDD nuestro problema principal es estar convencidos de que llegamos a suficientes neuronas, porque los ratones tienen un cerebro chiquitito que es fácil llenarlo entero con el virus pero nosotros tenemos un cerebro mucho más grande (un niño de 5 años tiene un cerebro más de dos mil quinientas veces mayor que un ratón). Por eso el desafío es llegar a tener una distribución del virus suficientemente amplia en monos y cerdos que tienen cerebros más medianos que estemos seguros de que al saltar a humanos consigamos suficientes neuronas infectadas para obtener eficacia.
Y hubo bastante debate en el Foro sobre cómo sabremos que es “suficiente” para poder decidir empezar ensayos, y la respuesta consenso es que debemos estar bastante seguros antes de tomar la decisión. Y aún parece que lo conseguiremos en el futuro cercano, pero por eso es tan difícil saber cuándo exactamente. Y el consejo de los expertos de industria es que en cualquier caso, aunque sepamos que el futuro nos traerá terapias mejores, eso no debe frenarlos a la hora de avanzar algunas de las del “casi ahora” a ensayos en cuanto estemos bastante seguros de que las sabemos usar lo mejor que podemos. Porque por un lado no podemos esperar para siempre, las personas con CDD no deben esperar muchos años sobretodo si son niños, y por otro lado porque la verdad es que no sabemos cómo de buenas serán esas terapias futuras ni cuando llegarán.
Y mientras escribo esto estoy pensando que por eso necesitamos meter más caña al desarrollo de las terapias dirigidas a la causa que no usan virus, como la hacer la proteína en laboratorio (de reemplazo enzimático) o los ASOs, porque los científicos no sabemos aún como dar una segunda dosis de virus a una persona que ya ha recibido una terapia génica con virus pero esas otras si son combinables. Así que espero el año que viene en el Foro oír de los progresos con esas terapias no víricas.
5. Cómo puede ayudar la comunidad de pacientes

Panel de expertos, de izquiera a derecha: Ralf Schmid (Novartis), Sharyl Fyffe-Maricich (Ultragenyx), Randall Kaye (Longboard), Brian Moseley (UCB) y el moderador Majid Jafar (Fundación Loulou)
Tuvimos un panel con expertos en desarrollo de fármacos y terapias génicas de industria para hablar de cómo desde la comunidad de pacientes podemos ayudar a acelerar el desarrollo de tratamientos. Algunos de sus mensajes fueron:
EDUCACIÓN/INVESTIGACIÓN: Ayudar a las familias a entender que participar en algún proyecto de investigación debería formar parte de lo normal para familias, por ser una enfermedad rara. No tiene por qué ser un ensayo clínico con fármacos, sino los estudios observacionales como CANDID o de biomarcadores como ELPIS. Si los pacientes no participan no podremos sacar ademánte la nueva generación de tratamientos.
INVESTIGACIÓN: Ayudar a validar escalas cognitivas, motoras y comportamentales para poder documentar en ensayos clínicos la eficacia de los tratamientos que no son anti-epilépticos (yo creo que esto ya lo hacemos bien con todos los estudios que hay en marcha).
EDUCACIÓN a familias sobre terapias génicas: qué se puede esperar realísticamente de una terapia de este tipo, cual es el proceso de participar en un ensayo clínico con una terapia génica…
POLÍTICA: hay desafíos que no podemos arreglar nosotros, pero que podemos influenciar, como es ayudar a que las agencias reguladoras entiendan la necesidad y el valor de una terapia génica para CDD, y también el problema de precios de terapias génicas.
INVESTIGACIÓN: También algo fuera de nuestro control pero donde podemos apoyar a investigadores y unir fuerzas con otras enfermedades es el tema de ver cómo hacer que las terapias génicas con virus se puedan dar más de una vez. Si supiéramos que hay una segunda oportunidad y no solo una, haría más fácil la participación en ensayos y también a las empresas hacer esos ensayos. Una forma en la que podemos ayudar en este campo es por ejemplo teniendo una sesión centrada en la respuesta inmunológica a la terapia génica en el próximo Foro, y en cómo poder evitarla para poder dar la terapia más de una vez.
Y quiero añadir dos áreas donde los grupos de pacientes pueden aportar mucho pero que no fueron mencionadas en el panel:
INVESTIGACIÓN / COMUNIDAD: En el Foro vimos que varias de las ponencias eran sobre proyectos de investigación que fueron empezados con fondos de los grupos de pacientes de ese país, como Canadá, Italia y España. Y eso nos demuestra que financiar aunque sea con poco la investigación es otro papel importante de los grupos de pacientes, que con el primer grano de arena comienzan la creación de una montaña. Por eso creo que aportar esos primeros granos de arena a que se investigue CDD en sus países es una labor importante de los grupos de pacientes.
COMUNIDAD: Necesitamos encontrar más pacientes. Está claro que hay más gente con CDD sin diagnóstico que con diagnóstico, y cada paciente sin diagnóstico es una persona que podría estar en el tratamiento equivocado, que no puede entrar en esos ensayos, y que no puede ayudarnos a responder todas estas dudas clínicas como la epidemiología verdadera del síndrome. Y creo que esto es un problema que hay que atacar país por país porque las barreras al diagnóstico son únicas de cada sitio, ¡así que nos lo pongo en la lista de tareas!
6. La voz de los pacientes: comunidad, esperanza, y manos a la obra
El Foro tradicionalmente lo abre y lo cierra una ponencia de familiares, y este año lo abrió Lily Howard que es la hermana de Harper y nos habló de su experiencia como hermana. Nos habló de la soledad, y de la madurez forzada, y de sentirse distinta a los otros niños e incapaz de conectar con ellos. Por eso Lily ha empezado un evento para hermanos donde puedan crear entre ellos la sensación de comunidad y pertenencia que les falta en su ambiente familiar y escolar.

Imagen: Majid y Lynn Jafar (Fundación Loulou, padres de Loulou); Lily Howard (Hope-4-Harper, hermana Harper); Andreas y Ana Born (CURE5; padres de Siena); y Heike Knip (Presidente de la CDKL5 Alliance y padre de Valentina)
Durante la cena de gala, el premio a los pacientes fue para Ana Carolina y Andreas Borg, padres de Siena, que nos hablaron del shock inicial con el diagnóstico y de descubrir un mundo que no sabían que existe, y de cómo encontraron su lucha por conseguir tratamientos para CDD tanto un propósito de vida como una sensación de tener agencia. Os comparto una frase de Andreas: “Todos los problemas solucionables tienen solución si suficiente gente con talento pone su energía en resolverlos” y una de Ana: “todos necesitamos un héroe, y yo estoy orgullosa de estar criando el mío”. Esta es la noche del congreso en el que todos acabamos llorando y confiando en que podemos construir un futuro mejor.
La última ponencia del Foro fue del actual presidente de la Alianza, Heike, quien nos habló de nuevo de la necesidad de estar unidos en la comunidad de pacienes y nos hizo un resumen de los logros de cada grupo nacional de pacientes de la alianza en este último año. También nos recordó que debemos buscar mejorar la calidad de vida de las familias, no solo centrarnos en la terapia génica, y nos daba el ejemplo de cómo su hija ha pasado de tener 3 o 4 crisis al día a tener 3 o 4 crisis a la semana en su últimos ensayo clínico, y cómo pada pequeña mejora ayuda muchísimo. Esto recuerda a cómo Lily nos dijo también en su ponencia de apertura que incluso las tecnologías de asistencia (de apoyo) pueden mejorar mucho la vida de los afectados y sus familias, no solo las terapias médicas. En ambos casos el mensaje es que no nos olvidemos de que estamos intentando ayudar a personas, no a genes.
Por último, Majid Jafar que es co-Fundador de la Fundación Loulou, daba broche final al Foro convocándonos a vernos en Roma en Junio de 2025 en la reunión de la Alianza y anunciando que el próximo Foro vuelve a Boston los días 27 y 28 de Octubre de 2025. Y nos dejó con una llamada a la acción: “hemos llegado muy lejos, pero nos queda mucho camino por recorrer. Manos a la obra”
Así que nos volvemos al trabajo con la cabeza llena de ideas y el corazón lleno de amigos. Os veo en Roma y en Boston el año que viene.
Ana Mingorance, PhD
Nota: este texto captura mis impresiones de las presentaciones del Foro que más me interesaron, no es un texto oficial del congreso emitido por la Fundación Loulou. Escribo estos resúmenes para los padres de personas con CDD, así que a veces me tomo ciertas licencias a la hora de explicar las partes mas técnicas.

DRAVET SYNDROME AND HOW WE CAN TREAT A HAPLOINSUFFICIENCY
Patients with Dravet syndrome caused by missense mutations in SCN1A are candidates for the clinical trials with ASOs and gene therapies to increase SCN1A expression, and will be candidate for this therapies after approval.
I have heard some confusion around patients with Dravet syndrome who have missense mutations in SCN1A: are they candidate for the genetic therapies currently in trials or are they excluded from the trials?
The short answer is “Patients with Dravet syndrome caused by missense mutations in SCN1A are candidates for the clinical trials with ASOs and gene therapies to increase SCN1A expression, and will be candidate for this therapies after approval”. The rest of this entry is the longer answer.
What is a haploinsufficiency
If you have a child with Dravet syndrome with a mutation in SCN1A, as most cases have, you might have heard the word “haploinsufficiency”. Haplo means “single” in greek, as opposed to couple or two, so haplo-insufficiency applied to genetics means that they have insufficient copies of that gene because they have a single one, as opposed to two functional copies which is the default for most genes.
In the case of Dravet syndrome caused by mutations in SCN1A, the person has one good gene copy and one bad copy (due to mutations), which leads to no functional protein being produced from that back copy. That leaves them with 50% of the number of good sodium channels Nav1.1 that their neurons need. They have only half of what is sufficient.
The reason I am writing this entry is that we often describe Dravet syndrome as a protein haploinsufficiency, as if patients only had 50% of the sodium channels. But this is a simplification that can worry families of patients with missense mutations who actually have 100% of the sodium channels…. But half of those don´t work.
We also often use mouse models for the disease where indeed only 50% of protein is produced because they have one good copy of SCN1A and one copy that makes no protein. So we think that this is what always happens, they produce only half of the numbers of sodium channels that they need. The consequence, by the way, is less sodium then running through the neuron membrane and less neuron firing. And it is very intuitive to understand that the gene therapies and other genetic treatments will try to bring expression “back to 100%” to help neurons function well.

We often explain Dravet syndrome as only having 50% of the Nav1.1 sodium channels
But what about missense mutations then?
There are many patients with Dravet syndrome that have a mutation of a type called “missense”, the one that doesn’t break the gene, but instead gives you a mistake in the sequence for the sodium channel protein, so that it will have one wrong letter. So you still probably produce 100% of the sodium channels, but only half of them works. The consequence is exactly the same as if the non-functional channels were not even there, it results in less sodium running through the neuron membrane and less neuron firing. But families and even some scientists worry that these patients cannot get the genetic treatments that increase expression of the gene because it will increase both the good and the bad copies. But it doesn’t matter in Dravet syndrome. The bad copies are not “bad” as in doing something bad, they are just useless. In science we call them “loss of function”, they really don’t work, they have lost their function.
I call these cases “functional haploinsufficiencies”. Yes they still produce channels from the bad copy, but these are no longer functional, so when it comes to functional sodium channels they still have half of the ones they needed.
I always understand better if it is in visual form. So I will adapt one of my old cartoons where I pictured the sodium channel as a door in the membrane that lets sodium come in. I am also guilty of only drawing the 50% of protein being produced scenario, which is true for mutations that break the gene and make no protein. But I never draw the missense scenario, where the number of doors for sodium is still fine but half of them are closed. The result is functionally the same: the neuron only has half of the doors that it needs to get enough sodium in. It doesn’t matter if the other half of the doors are missing or closed. It really doesn’t matter.

Missense mutations still result in only half of the sodium channels (the sodium doors) being functional
For me the door visual makes it much easier to understand why the different types of mutation result in the same biological protein. It is like everyone trying to leave a concert at once and having only half of the doors that could handle that many people. It doesn’t matter if the other half of the doors don’t exit or are shut, the result is the same, not enough usable doors.
By the way years ago we made a Dravet mouse model with a missense mutation that has been seen in several patients. And we then looked at what happened to that channel: it is produce and nicely sits in the neuron membrane as it should, but it remains closed at voltages where the good channels open to let sodium in. In door terms, there is no problem in the number of doors, the mutant ones are just closed all the time, and therefore as useless as if they were missing.
Why is this important for clinical trials?
There are currently two therapies in clinical trials for Dravet syndrome that are designed to increase expression of the sodium channel Nav1.1 by increasing the production of protein (the channel) from the SCN1A gene.
The most advanced therapy does this by using a small piece of RNA called an “antisense oligonucleotide” or ASO for short. It is the drug zorevunersen from Stoke Therapeutics (also known as STK-001). The second one is a gene therapy that uses a virus to bring to neurons the instructions to read the SCN1A gene more. It is called ETX101, from Encoded Therapeutics. I won’t get into the science details, you just need to know that both are currently in clinical trials for patients with Dravet syndrome caused by mutations in SCN1A, and both work by ending up in an increase of good sodium channels (opened doors at the concert).
Both clinical trials accept patients with the two types of loss of function mutations in SCN1A, including the missense. They are not only for patients that have mutations that break the SCN1A gene (nonsense, frameshift, gene deletions). That is a misunderstanding that worries families and the reason for this post.
ETX101 trials asks for “Participant must have a predicted loss of function pathogenic or likely pathogenic SCN1A variant.”
Zorevunersen trials asks for “Documented pathogenic, likely pathogenic variant, or variant of uncertain significance in the SCN1A gene associated with DS”. By the way this trial is not currently enrolling, they finished their Phase 2 trials and are preparing for the Phase 3.
Also VERY important, the large Phase 1/2 trial with zorevunersen is completed, showed safety and efficacy in the patients treated with the genetic drug, and the company has publicly explained that even though they don’t have enough patients to mathematically compare missense versus the other mutations, there doesn’t seem to be any signal that the missense patients don’t respond to this treatment. In this trial they had 45% of patients with missense mutations, and 55% with truncating mutations which are the ones that make no protein from that gene copy.
So it is not true that clinical trials to upregulate expression of SCN1A are only for patients with truncation mutations. But what about safety?
Another worry by scientists has been that the brain won’t handle having 200% of sodium channels (of doors), that it will be dangerous. But the science to support this belief is not there, and in fact both companies had to evaluate their genetic therapies in healthy non-human primates (monkeys) who don’t have Dravet syndrome and therefore have their two GOOD copies of SCN1A, and to show that their therapies increase the production of sodium channels and the monkeys are still fine. Companies need to prove this before getting permission to run trials in patients. So monkeys are fine with 150-200% or more of open doors (letting more sodium in!), which means that we don’t have worries for patients going from 50% of open doors to hopefully 100% of open doors. The extra closed doors, if they have missense mutations of the type that results in protein being made but not functional, are really not a problem.

Gene therapies and ASOs that increase expression from the SCN1A gene or mRNA are also in trials for patients with missense mutations in SCN1A
IN CONCLUSSION
Patients with Dravet syndrome caused by missense mutations in SCN1A are candidates for the clinical trials with ASOs and gene therapies to increase SCN1A expression, and will be candidate for this therapies after approval.

What I learnt at the SYNGAP Conference 2023
These are my main learnings from the Syngap Conference 2023 celebrated in Orlando the day before the American Epilepsy Society meeting.

Last year I attended the Syngap Conference organized by the SynGAP Research Fund the day before the American Epilepsy Society (AES) meeting. It was fantastic, but I did not write a separate summary for it, only a mention as part of my AES summary. But then this year I attended again the Syngap Conference, again the day before AES 2023 started, and I believe it deserves a separate summary. So here it goes: these are my main learnings from the 2023 Syngap Conference about the SYNGAP research and therapeutic field.
1 – COOL AND SCARY NEW BIOLOGY

There was a lot about the biology of the SYNGAP1 gene and the SynGAP protein at the conference. The main learnings that stayed with me are two:
Did you know that SynGAP seems to control cytoskeleton (the cell skeleton) and in particular the primary cilia? This is like a special part of the neuron that looks like a short tail. This is interesting because I work very much in CDKL5 deficiency disorder (another neurodevelopmental syndrome with epilepsy) and CDKL5 is also doing the same things!! This whole “cilia connection” is very cool and also very puzzling. I don’t know what to do with this information, but it seems that proteins that evolved to control the cell skeleton and that special primary cilia might have also evolved to control synaptic remodeling and that is why we get neurodevelopmental syndromes with epilepsy when they are mutated. Helen Willsey from UCSF was such a great speaker! One of the highlights of the conference.
That was the cool new biology. The scary new biology is that we learnt that syngapians spend in REM sleep only half of the time that other people do. REM sleep is when we dream… so having SYNGAP steals the dreams of the kids, and this is very sad. The good news is that REM sleep could be a biomarker for these kids.
2 – HELP OUR KIDS NOW: DRUG REPURPOSING
There were several talks about drug repurposing, which means seeing if there could be already something sitting at the pharmacy shelf (perhaps even in the supplement shelves) that could help syngapians today. Repurposing is the search for faster treatments while investing in the long term disease-modifying treatments.
We learnt about a research program by Rarebase using patient-derived neurons to characterize some potential drugs that may help increase the mRNA of SYNGAP1 (remember that there is one good copy of the gene that we can use to get more protein), and one in fruit flies missing SYNGAP1. The mutant flies have small eyes looking disorganized, which serves a read out to look for drugs and supplements that could fix their problem and therefore compensate for lack of SYNGAP1. The researcher, Clement Chow, explained that they are finding many drugs that come up as positive and that have similar action, like many anti-inflammatory drugs, so the results are looking promising. And we also heard from Zach Grinspan who has been running a repurposing trial in SLC6A1 and STXBP1 with a drug called phenylbutyrate that will also be tested in SYNGAP. This talk opened a very interesting debate about this type of repurposing trial. For example: do we need a consensus to pick the winner compounds? How are we going to test the potential repurposing drugs if they are already available for example at CVS? Should we go through 6 months of paperwork just to have ethical approval and spend much money on trials or go ahead and try it individually? And how would we learn from those N-of-1 family-run studies?
Prof Ingrid Scheffer, who is one of the most famous doctors in the rare epilepsy space, insisted that the only way to know if something works (and is safe) is to run randomized controlled trials, and that the burden should be in the companies that own those drugs, not on the patient groups. This was a great debate!
3 – THERAPIES TOMORROW: HIGHWAY TO CURES
To cure a disease like SYNGAP we know very well what we need to do: help neurons make more SynGAP. I moderated this session with the message that “we have all of the ingredients that we need to fix SYNGAP”: we know the cause, we have one good copy of the gene to use, the disease is not degenerative, the animal models are good, and the animal models indicate that the disease is likely reversible. We are also talking about a relatively large rare disease, with a very strong patient community working on solving the right problems, and there are already several companies working on treatments.

We had a presentation from Praxis and one from Stoke, both working on ASOs to boost SynGAP production using the good gene copy. In both cases, these companies have more advanced ASO programs for other epilepsy syndromes so we will get to use those learnings for SYNGAP. And we also had a presentation from Tevard, a company working on a gene therapy to help neurons skip premature stop-codons caused my non-sense mutations. Tevard is planning to run clinical trials with this gene therapy in a selection of genetic epilepsy syndromes including SYNGAP, and they are ready to start safety studies in non-human primates next year to then progress to clinical trials.
I also know that there are more therapies being developed to target the cause of the disease, and some of those companies where present in the room although they didn’t give a talk, so therapies are coming.
And to prepare the field for those therapies the community is running A LOT of clinical studies to know the natural history of the disease and possible outcome measures (scales) to use for trials. A LOT. This included a disease concept model, the Ciitizen study, and an excellent endpoint-enabling study at CHOP that is designed the same way that companies design their observational studies, this is top quality. Also a lot on biomarkers. What I wonder is how to integrate all these data. What is clear is that with so much work this is not a road to trials, but a highway to trials, in particular the trials for disease-targeted treatments (the cures).
4 – THE KEY CHALLENGE OF MISSENSE CAN’T WAIT
The disease that we often call SYNGAP for short happens when one of the two copies of the SYNGAP1 gene is mutated, and this mutation sometimes breaks the future protein (non-sense, frame-shift, truncation, deletions) but sometimes the mutation makes a protein that has one wrong letter. This is not trivial, and read this section until the end to know the mind-blowing implications. This means that many SYNGAP patients have a mutated protein instead of a missing protein. What if we can fix those? Maybe they just need a bit of help with folding. What if they stay around and don’t let the good proteins work? Then we need a strategy to remove them. This is why there are several programs trying to understand what happens to those mutated proteins, which are often unique to each patient with a missense mutation. This includes in silico modeling and also in vitro real experiments.
But the biggest implication is perhaps not for treatment, but for diagnostics. We learnt from Gemma Carvill that genetic labs often don’t know how to interpret these changes of letter, so patients receive a genetic report for “VUS” or Variant of Unknown Significance. With a VUS you cannot be diagnosed, because maybe the gene is totally fine (we all have some different letters here and there in our DNA that are harmless). In SYNGAP1 gene sequencing for patients with epilepsy, the large majority are VUS, like 80% or so, so they don’t get a diagnosis. The other 20% is split between benign (trivial) mutations, and pathogenic which are the real SYNGAP. But then Gemma and her colleagues train a computational model to predict if a missense is bad or trivial, they found out that about 22% of the VUS are likely pathogenic mutations. What this means is that if we could solve the meaning of the missense mutations, the diagnosed patient population with SYNGAP could multiply by 3 or so. This has mind-blowing implications, not only to grow the patient population but to then help those new patients access better treatments and clinical trials and a community of families like theirs.
No wonder this is such an important priority for the SynGAP Research Fund!
5 – ALL FOR ONE
A real team
I am used to seeing patient groups focused on one country or territory, even when they call themselves international. This is understandable. I am a Spanish scientist, so I often collaborate with the groups in Europe (proximity and shared regulatory framework) and in Latin America (shared language and culture). And what I love about the SynGAP Research Fund is that it integrates all those worlds. You could even see that in the logos in all the conference materials:
SynGAP Research Fund
SynGAP Research Fund EU
Fondo de Investigación de SynGAP
And you could also see it around the room, because this group is very international. We had Mike and Aaron and Hans and Sidney and Lauren and others from the US, and Vicky and Martha and Carlos Caparrós and others from Latin America, and Katrien and Virginie and likely others from Europe. This is a true international team and this is exceptional and wornderful.
Get treatment for everyone
A message that was heard multiple times during the scientific program was the concern for all families living with a syngapian:
The focus on repurposing was: how can we get these treatments to everyone?
The focus on missense was: how can we find all those missing families so they are not left in the dark?
The joint work of the different territories US/EU/LATAM is also about getting to everyone.
Mike Ganglia closed the scientific conference with the words “what are all the people out there doing with their non-diagnosed syngapians?! We owe it to those parents”
A unique community
Clement Chow (the fruit fly scientist) explained that “Mike found us on Twitter, where all beautiful relationships start”, referring to Mike Graglia. And now Clement is working on finding repurposing candidates for SYNGAP!
My own path to get to know this community was similar. I was invited to give a talk in San Diego in 2018, and a SYNGAP dad reached out to me also through Twitter and asked me if I would like to meet his son with SYNGAP and his family while I was in town. That was Aaron, Jaxon’s dad, and it is a very good strategy to get scientists to get interested in a rare disease.
These two stories are an example of the proactivity of the SRF and the SYNGAP patient community, and other quotes that I wrote down during the 2023 conference also communicate this same message:
Tom Frazier, who is developing web-based measures of behavior and cognition: “This community volunteers for studies more than any other”.
Clement Chow: “[…]the strength of foundations leading the therapeutic discovery for rare diseases right now, and this foundational machine is best oiled than any other foundation that I have worked with, you should be proud of it”.
Then the following day was the Families Day with I believe more than 40 families attending and even using the opportunity to collect clinical data for some of the research studies. This community is exceptional and ready to partner and drive research forward.
* * * * * *
I hope you enjoyed this summary. If you are a SYNGAP family member, try to join us next year in Los Angeles right before AES 2024, there is also a second day focused on families after the scientific day. And if you are a scientist or clinician, please also come to LA one day early. This disease has all of the ingredients that we need to fix it, and a unique community behind it. Please join.
PS: my summary from AES 2023 is HERE

TOP 5 INSIGHTS FROM THE AMERICAN EPILEPSY SOCIETY MEETING 2023
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. These are my top 5 insights from the American Epilepsy Society 2023 meeting.
I often write a summary of the main lessons from the American Epilepsy Society meeting, but for a second year there was so much about rare epilepsy syndromes or DEEs that I was not able to follow the main conference. I only went to one lecture session, and focused instead on the posters, scientific exhibits and meetings, which are all the opportunities to directly talk to people and by far the main value of this congress.
This is my summary of the trends that I am seeing in rare epilepsy syndromes after attending AES 2023. It is not a summary of the conference itself, which has really become too large to cover. In particular, I wanted to see if 2023 delivered on the promise set last year, which was Escape velocity for genetic epilepsy syndromes.
1 – EPILEPSY AS A COLLECTION OF RARE DISEASES
The larger pharma space has experienced a clear shift towards more personalized medicine and more orphan drugs. And epilepsy is not an exception. Since around 2014 with the clinical trials of cannabidiol, we have seen an explosion of programs directed to epilepsy syndromes, both with symptomatic and with gene-targeted approaches.
At AES 2023 I attended the plenary session where Dr Kelly Knupp from Children’s Hospital Colorado showed one slide with a key title, which ended up getting very popular on social media. It read: “Epilepsy is a collection of Rare Diseases”.
I believe this probably holds true for all medical fields, where as we gain insights into the cause (etiology) of diseases we realize that one large disease is actually many orphan-size diseases all sharing some common phenotype. But it is important to talk about this, and it was important to hear this message in the large session, because it captures where much of the attention right now is going within the epilepsy space: to the rare diseases.
And this was particularly visible at AES 2023, where many of the pharma stands at the exhibit floor and pharma-sponsored sessions focused on rare epilepsy syndromes. As last year, this was largely focused on the most famous syndromes: Dravet syndrome, LGS and TSC; and this year also included CDD, the 4th rare epilepsy syndrome with an approved treatment, and the subject of an additional Phase 3 trial by UCB pharma.
Yet the poster session gave us a glimpse into the future, and I would like to highlight a poster from Biogen on KCNT1 and two from UCB on STXBP1 and SLC6A1. Although all preclinical, these posters are important because they illustrate how the large companies are now proactively working on the genetic epilepsy syndromes, not just watching the field (as they did at first) or buying the orphan drugs developed by others (more recently). This is an important step for the industry.
With large biotech and pharma now on board with genetic epilepsy syndromes, we are reaching a more mature stage in that transition from only working on seizures to also working on genetic causes. And this is happening without taking attention away from the larger seizure treatment space, where I would highlight the impressive results with XEN1101 so far in focal-onset seizures and potential also for generalized seizures. This is a very good scientific time for the epilepsy drug treatment space in general.
2 – RARE EPILEPSIES AS A TRANSITION POINT IN THERAPY DEVELOPMENT IN EPILEPSY
Are rare epilepsy syndromes the future of epilepsy treatment? Not at all. The syndromes are rather a natural step in the transition from symptomatic treatments to treatments that treat the cause of the disease.
I really liked a presentation by Dr Gemma Carvill from Northwestern about the history of epilepsy. She highlighted how most of the progress so far has been not on efficacy (we always hit that 30-35% of refractory patients) but on having better tolerability, and how more recent development focused on rare epilepsy syndromes, then on genetic treatments for these syndromes, and how the future direction is to use those technologies to treat epilepsy at large. When applied to the non-monogenetic epilepsies, the main approaches would be gene regulation approaches, moving up or down the expression of important targets, instead of activating or inhibiting proteins as small molecules often do.

Important advances to modulate gene expression in epilepsy are dCas9 approaches (for example Colasante 2020), the use of activity-dependent promoters (for example Qiu 2022), or the targeting of non-coding RNAs (reviewed in Stine 2023). And one of the best examples of going beyond the rare epilepsies is the AMT-260 gene therapy program from uniQure for Temporal Lobe Epilepsy. Gene therapy trials are not only for monogenetic epilepsy!
So genetic epilepsy syndromes are not the ultimate destination, but the first application of a new way of treating epilepsy, and this is becoming more clear as the epilepsy field evolves.
3 – PRESSURE IS HIGH, ALL SYNDROMES NEED BETTER TREATMENTS
Dravet syndrome is one of the epilepsy syndromes that has received the most attention from the industry. It has three drugs approved, soticlestat in phase 3 trials, and is the target of the most advanced clinical trial with a therapy designed to correct the cause of an epileptic syndrome: the ASO STK-001 from Stoke to correct the haploinsufficiency in SCN1A. This makes Dravet syndrome the “lucky syndrome”, and I have even seen grant proposals for Dravet turned down because it was considered “already done” by reviewers.
Yet at the Dravet syndrome roundtable we got to hear about 6-year old Anna and her journey with Dravet syndrome from her grandpa Ted Odlaug, who is the current president of the US Dravet Syndrome Foundation. During her short life, Anna has tried multitude of anti-epileptic drugs, including the three approved for Dravet syndrome, yet her epilepsy has gotten worse and worse and she currently has no days without seizures. She also has the cognitive development of a 2-year old, limited vocabulary, and her high seizure frequency including nocturnal seizures place her at very high risk for sudden death (SUDEP). Yet she got the “lucky syndrome”, the one with the most advances towards treating the symptoms and the cause.
This is something important to hear and to remember. All of what we have in epilepsy syndromes are some treatments approved to treat seizures, which work in SOME patients and that are tolerable by SOME patients. The treatments to correct the cause of these diseases are still in early development and still only accessible in the context of clinical trials. And this holds true even for the “lucky syndrome”. Most other syndromes are still behind in terms of therapy development.
So there is a big gap between the science, where we have cured the mice with SCN1A deficiency with several gene therapies and ASOs, and what patients in the real world like Anna have access to. Why is there such a progress gap between science and patient care? Part of it is time, treatments simply take years to get from the lab to the market, but we are also facing challenges bigger than time. And we are seeing these additional challenges as the first disease-targeting programs make their way to clinical trials and progress slows down. I discuss this in the next section. For this section my main message is that all syndromes need better treatments, and that none of them is yet “done”.
4 – RARE DISEASE THERAPY DEVELOPMENT HAS THREE MOUNTAINS TO CLIMB
Do you know the metaphor of a climber struggling to climb a mountain only to reach the summit and realize that there are other mountains just as high behind it? I believe this is where we are right now in developing disease-targeting treatments for genetic epilepsy syndromes, we are getting to the summit of that first mountain and looking at the next two. It turns out that getting gene-targeted treatment to trials was just the first mountain.
We had many discussions about these challenges during AES 2023. In fact, this was probably the most common topic across my many discussions with other attendees.
Mountain 1: Science.
Science has been advancing quite well. There are many preclinical programs with gene therapies and ASOs ongoing for a growing number of epilepsy syndromes, and we start having clinical trials. This year we have seen newer data from Stoke showing that with the highest dose of STK-001 they can have profound seizure reduction in patients with Dravet syndrome, and that even with moderate doses they can see improvements in non-seizure domains like communication. And Praxis has also started clinical trials with another ASO, PRAX-222, this one to reduce SCN2A in patients with gain-of-function mutations. This is in addition to Praxis’ small molecule trial for SCN2A and SCN8A gain-of-function DEEs.
We have also seen this year hopeful clinical trials results in Angelman syndrome (Ultragenyx) and Rett syndrome (Taysha) that make us all very hopeful that the neurodevelopmental field is getting closer to true disease-modifying treatments. So overall we start seeing the transition from preclinical ASO and gene therapies to clinical trials. This is just the first mountain.
Mountain 2: Regulatory.
This is a difficult one. We are still not sure how to design a clinical trial to show the difference between correcting the gene versus treating the seizures, and there are no precedents for a broader label to treat the disease (all we have are approvals “for the treatment of seizures in syndrome X”).
The patient community, clinicians and the companies developing treatments to target the cause of the epilepsy syndromes are working to solve this challenge by running observational clinical trials to validate non-seizure outcomes and endpoints.
The most advanced studies are the ones on Dravet syndrome by Stoke and Encoded, that have documented how, for example, the number of seizures in Dravet syndrome remains constant over time (up to 2 years!) as a group average. So while one patient might get better or worse, if you run a trial the number of seizures should balance out and remain stable. Many other symptoms are also stable or improve just a little, which will help us interpret open-label studies that may show improvements. And there are also other endpoint-enabling observational studies going on for CDD, SYNGAP and STXBP1 (at least) that I know.
We still don’t know which of these scales will be useful in clinical trials to show improvements beyond seizures, or how to convert the scales into specific endpoints, but in the meantime some of the companies with approved drugs to treat seizures in DEEs are leaning quite far into almost claiming efficacy in non-seizure outcomes by relying on caregiver surveys. And this is a development that worries me. We already have the problem in rare diseases of having to overrely on case studies due to so few placebo-controlled trials. And now we are adding the problem of companies with anti-seizure medications using caregiver surveys not reviewed by regulators to talk about improvements in behavior and cognition. This was particularly visible in 2023, and it almost creates two tracks with different burden of proof for non-seizure outcomes: one of post-approval caregiver surveys for small molecule anti-seizure drugs, and one of years of endpoint-enabling studies followed by year-long trials with not-yet-validated outcome measures for ASO and gene therapies. I’m not liking this development.
Mountain 3: Market.
A common challenge for rare diseases is not having enough patient numbers for an attractive return-on-investment. We have seen this addressed in rare epilepsy syndromes by developing the same molecule for a group of rare diseases. For example Epidiolex approved for Dravet, LGS and TSC; Fintepla approved for Dravet, LGS and in Phase 3 trials for CDD; and soticlestat in Phase 3 trials for Dravet and LGS. But running multiple parallel trials is expensive, so companies focus only on the largest syndromes.
A near-future regulatory innovation might be clustering syndromes into a broader label for drug development. This year we heard from the company Longobard, with a Phase 2 basket trials in DEEs, that they will consider discussing with regulators the potential to keep the combined DEE target population for Phase 3 trials and approval. I see regulatory challenges to do this but I also see it as the only way to get clinical trial data for many of the DEEs beyond the top 6 or so in terms of patient numbers, so I would love to see this happen.
The company Tevard is also considering a combination of DEEs, in their case because they are developing a gene therapy to help the cell skip premature stop codons caused by non-sense mutations, so their case is very similar to basket trials in the cancer field. In the case of Tevard, the target population would be genetic DEEs caused by non-sense mutations, and the common primary endpoint could be reduction in seizure frequency so one trial could test them all. Even a clinician working for Jazz told me that they had been asked by many clinicians to run studies with Epidiolex in groups of biologically-related DEEs (for example ion channels, or synaptopathies) so this definitely seems to be a conversation that we are hearing loudly in 2023 and that will likely turn into action.
5 – IMPRESSIVE PATIENT GROUPS
I have been working on rare epilepsies and supporting patient groups for 12 years now. And oh boy has that changed. Patient advocacy in 2023 is much more advanced and professional than how it was back in 2011.

The four epilepsy syndromes that have drugs approved all have very impressive patient-run foundations and patient groups behind them. See for example how the AES 2023 Extraordinary Contribution Award went to Mary Anne Meskis, Executive Director of the Dravet Syndrome Foundation. But they are no exception anymore. During AES 2023 I had a chance to interact with some of the patient groups for syndromes that still have no treatment approved (or advanced clinical trials) that were equally impressive.
Some of these deserve a shout out:
I attended the SYNGAP conference by the SynGAP Research Fund the day before AES. I wrote a separate summary about this conference, but this group is amazing in how they are tackling the key challenges to therapy development in their field, scientific/medical/industry community building, and patient mobilization including a very international footprint.
I caught the end of a meeting between the FamilieSCN2A Foundation and a biotech company, since I was next to meet with the company, and I was very impressed with their print slide-deck summarizing all of the resources and learnings from the SCN2A field. This included the entire list of mouse models and efforts to also validate outcome measures, for example in communication. This will help companies get up to speed with SCN2A quickly, and I’m pretty sure it will help start treatment programs by making it easier to choose this syndrome.
It is a tradition for me to sit down with Charlene and John from the STXBP1 Foundation every year at AES and compare notes about their field and the syndromes that I am working on. This year Charlene showed me one slide with the STXBP1 pipeline in 2019, having only one program for potential repurposing with nothing else on the horizon. And then the 2023 pipeline slide, with 11 programs in development including several gene therapies being developed by companies. Talk about progress!
CONCLUSSION: HAVE WE REACHED ESCAPE VELOCITY?
In science: yes. We are seeing a growing number of ASOs and gene therapies being developed for several of the neurodevelopmental syndromes with epilepsy. Transition to clinical trials is still a bit slow but the progress across many diseases is notable.
In regulatory path and market conditions: no. We are still not sure how to design a clinical trial to show the difference between correcting the gene versus treating the seizures, there are no precedents for a label that goes beyond seizures, and the market is still a limitation, leading to companies always choosing the largest syndromes and neglecting the other hundred or more.
We are seeing some development to address these challenges. Some are good developments, like considering how to get several syndromes into a single clinical program and approval. Some are (as I see them) bad developments, like companies promoting cognitive and behavioral efficacy for their drugs using a language identical to the one they use for seizure efficacy, which is the only one approved and on label.
Another development is the arrival of gene therapies to non-genetic epilepsy trials, which is very promising. Also exciting is to see big bio and big pharma proactively working on the genetic epilepsy syndromes, not just watching the field or buying the orphan drugs developed by others. And we are also now getting the results of the observational studies that are being carried out for several DEEs to identify endpoints for more complex clinical trials. So overall this was a good year.
For 2024 my hope is to:
see some answers to the regulatory challenges, with learnings from Stoke and also from other companies in the neurodevelopmental (non-epilepsy) field.
see more clinical trials started with treatments targeting the cause of the disease (potentially from the group of SLC6A1, SCN2A Loss-of-function, and CDD?)
see more mouse proof-of-concept data with treatments targeting the cause of the disease. We have many ideas and in vitro data, but not enough clinical candidates yet.
And I hope to see that in Los Angeles, for AES 2024.
Ana Mingorance, PhD
Disclaimer: These are my own impressions from the presentations and topics that I was most interested in. I write these texts with the parents of individuals with rare epilepsies in mind, so excuse also my lack of technical accuracy in parts.

REPASO DEL FORO CDKL5 2023
La novena edición del Foro CDKL5 tuvo lugar en Boston, los días 6 y 7 de noviembre. El Foro es una reunión anual que organiza la Fundación Loulou y en la que científicos y miembros de la industria farmacéutica se reúnen con representantes de la comunidad de pacientes para repasar los últimos avances en el campo.
Este es un repaso para los grupos de pacientes de las principales novedades del Foro CDKL5 2023.

FORO 2023
Hace ya nueve años que la Fundación Loulou organiza una reunión anual, el Foro CDKL5, donde los científicos de academia y de industria trabajando en el síndrome de deficiencia en CDKL5 (CDD), junto con representantes de los grupos de pacientes, se reúnen para compartir las últimas novedades y avanzar hacia tratamientos y una cura. Tenéis el resumen del año pasado aquí.
La edición de 2023 tuvo lugar en Londres los días 6 y 7 de noviembre, y volverá a Boston en 2024. Cada año digo que este ha sido el mejor Foro, pero es que es verdad. Tengo 47 páginas de notas por toda la información nueva y todos los momentos importantes que nos dio el Foro. Así que voy a intentar resumir las conclusiones principales de la edición de este año, no incluyendo todas las presentaciones sino centrándome en los temas principales y los progresos que vimos, que son muchos.
1. SEGUIMOS DESCUBRIENDO COSAS SOBRE CDD
El Foro siempre arranca y cierra con la voz de la comunidad de pacientes. Este año fue Cristina, de la Asociación de Afectados CDKL5, quién abrió el Foro con un mensaje muy poderoso. Nos explicó que cuando les llegó el diagnóstico de su hija, aprendieron que “viviría una vida a medio camino entre incierta y horrible”. Y que por eso, para allá, la investigación es la esperanza.
Y cada año la investigación nos sigue enseñando cosas nuevas sobre CDD que tienen importantes aplicaciones de cada a entender la enfermedad o a posibles tratamientos. Aquí os cuento algunas, y mirad también la sección 6 de este resumen.
La Dra Lauren Orefice de Boston nos enseñó como el rechazo a ciertas texturas en CDD es consecuencia de cambios sensoriales, no de comportamiento. Los ratones con CDD tienen las neuronas sensoriales demasiado sensibles a las texturas rugosas, reaccionan demasiado fuerte, y por eso suelen rechazar esas texturas. Y es muy posible que lo mismo pase en las personas con CDD que insisten en comer texturas específicas.
Varios proyectos de varios laboratorios coinciden en ver cambios de conectividad funcional en CDD, pero no cambios estructurales. O sea que las neuronas están en el sitio correcto y en general el cerebro está bien y bien conectado, pero algunas neuronas disparan juntas más de lo que deberían, y otras no disparan juntas tanto como deberían (eso es la conectividad funcional). Y para mi esto es muy prometedor de cara a tratamientos porque cuando normalizamos la actividad neuronal podemos obtener cambios en este tipo de conectividad funcional. Lo que sería mucho más difícil de arreglar es cambiar una neurona de sitio.
Sir Adrian Bird nos dio una de las ponencias estrella. Se hizo famoso porque fue el primer científico que demostró que el síndrome de Rett es reversible en ratones, y n os explicaba que en Rett (gen MECP2) tener poco o tener mucho es problemático, con lo que todas las terapias génicas en desarrollo para Rett (que es por falta de MECP2) llevan mecanismos de freno para no llegar a causar sobreexpresión (demasiado MECP2). Pues una de las sorpresas de este año nos vino de la Dra Sharyl Fyffe-Maricich de Ultragenyx, que nos demostró algo que sospechábamos desde hace algunos años: el cuerpo ya pone freno a cuanto CDKL5 puedes producir y no te deja hacer sobreexpresión. Nos enseñó datos con ratones y con primates, donde da igual cuanta terapia génica les des, las neuronas se comen el exceso de CDKL5. Y eso es buena noticia porque las terapias génicas de CDKL5 serán más fáciles al no tener que incorporarles ningún mecanismo artificial de freno.

Rechazando texturas y aprendiendo de Rett
2. ELIMINANDO BARRERAS PARA AVANZAR EL DIAGNÓSTICO Y LA INVESTIGACIÓN
El Foro 2023 nos trajo muchos ejemplos de generación de modelos de forma colaborativa, y quiero destacar un progreso en torno a diagnóstico y uno sobre modelos de epilepsia.
Un diagnostico genético no siempre es fácil de interpretar. Por ejemplo, si vuestro hijo o hija tiene una mutación en el gen CDKL5 que le rompe el gen, es fácil de interpretar en un test genético. Pero si tiene las de una letra cambiada por otra… esas son difíciles de interpretar y acabamos con un VUS (de las siglas en inglés para “variante de significado incierto”) y con un diagnóstico sin confirmar. Un equipo de la Telethon en Italia está haciendo en el laboratorio todas las mutaciones de cambio de letra posibles en CDKL5, probándolas en células, y van a poner todos los resultados en una base de datos. De ese modo, cualquier laboratorio genético encontrará respuesta para cualquier cambio de letra que vean en un niño. No más VUS en CDKL5. Un proyecto super chulo.
Y otro progreso enrome en generación de modelos de investigación que vimos este año es que por fin tenemos buenos modelos de epilepsia! La Dra Liz Buttermore nos enseñó un modelo in vitro, a partir de neuronas de pacientes en placas de cultivo, donde las neuronas de pacientes tienen una hiperactividad clara (disparan mucho). Y como científica que trabajó en la industria farmacéutica os puedo decir que este es el tipo de modelo que me hubiera gustado tener para probar fármacos. Luego está si recordáis el Dr Muotri de California que hace organoides (bolitas de neuronas) a partir de células de pacientes, que son como bolitas de corteza cerebral. Pues este año la Dra Rebeca Blanch, de su laboratorio, nos enseñó como las van a hacer tálamo-corticales para que puedan tener ese circuito tan importante para la epilepsia. Y el Profesor Peter Kind nos enseñó cómo su laboratorio ha encontrado el foco epiléptico en el cerebro de ratas con CDD, y ahora pueden estimularlo directamente para provocarles crisis cada vez que quieren. Este es un modelo fantástico para estudiar tratamientos. Sin duda en este año hemos tenido un avance tremendo en esta área.

Deshaciendo VUS y epilepsia en ratas con CDD
3. LOS “4 APROBADOS” Y LOS FÁRMACOS EN DESARROLLO
En 2022 se aprobó ganaxolona en Estados Unidos y se convirtió en el primer fármaco aprobado para CDD. Luego en 2023 nos ha llegado la aprobación Europea.
Y para que veáis lo excepcional y lo difícil que es tener un fármaco aprobado para un síndrome con epilepsia, os voy a repetir lo que dije durante mi charla en la sesión sorbe tratamientos: hay más de 300 síndromes con epilepsia, y solo 4 tienen un tratamiento aprobado. Estamos hablando de Lennox-Gastaut, esclerosis tuberosa, síndrome de Dravet y CDD. Ya está, solo esos 4.
Y una empresa de biotecnología empezó a llamarlos este año “los 4 aprobados”, por ser la excepción. Y para las empresas de desarrollo de tratamientos, estos 4 aprobados son más fáciles que cualquier otro, porque ya existe un precedente y por tanto respuestas a muchas preguntas de cómo llegar a la meta en esa enfermedad, y eso los hace más atractivos para tener más tratamientos. Por eso el valor de ganaxolona va más allá que el valor como medicamento.
Hemos pasado de haber tenido cero ensayos clínicos, a tener 8 en 8 años, y formar parte de “los 4 aprobados”. Para ser una enfermedad relativamente nueva, hemos empezado muy fuerte.
Y ahora la pregunta que nos queda es “y después de ganaxolona qué”. Y en el Foro vimos varias presentaciones de las terapias que vienen después, y que resume en esta sección y las tres siguientes.
Marinus anunciaba en el Foro que acaban de firmar un Programa de Acceso Global para llevar ganaxolona a los países donde no está disponible comercialmente (o en proceso de estar disponible como sería el caso de Europa). Su web tiene un correo de contacto para que los médicos puedan informarse. También nos enseñaron datos de que ganaxolona mantiene eficacia durante al menos 2 años.
UCB Pharma estuvo representada por dos personas, incluida la directora médica que dirige todos los ensayos clínicos, y nos explicaban que el ensayo clínico de fenfluramina para CDD en fase 3 está en marcha en Asia, Europa y Estados Unidos, con una web muy fácil de recordar: cddstudy.com
También repasamos varios fármacos en desarrollo para otras epilepsias o para otras enfermedades de neurodesarrollo, y que podrían potencialmente incorporarse a la lista de ensayos para CDD. Esos incluyen otros fármacos que actúan sobre el GABA, los activadores de KCC2, los inhibidores de Cav2.3 (os cuento más en la sección 6) y algunos tratamientos dirigidos a la flora intestinal que me resultan muy interesantes.
La conclusión de esta sesión es que ganaxolona fue solo el principio y que hay muchas opciones por detrás.

Presentaciones de empresas con terapias en desarrollo
4. CLINICAL TRIAL READINESS: CÓMO LA COMUNIDAD DE PACIENTES ESTÁ HACIENDO POSIBLES LAS TERAPIAS DEL FUTURO
Al terminal el Foro nos han llegado correos de gene de empresas que vinieron a Londres y todos destacan esta sesión como muy importante. Como nos escribió uno literalmente “esto va a atraer la participación de más empresas”.
Se trata de la sesión sobre biomarcadores y escalas clínicas, que responden la pregunta de “cómo vamos a medir en ensayos todo lo que no es epilepsia”. Y es particularmente crítico para los ensayos con terapias génicas, y en los últimos 12 meses hemos tenido muchos avances importantes.
Los biomarcadores son formas de medir cambios en el cerebro sin tener que hacer una biopsia (por razones obvias algo no viable para el cerebro). Un tipo de biomarcadores son los que podemos medir en sangre (los niveles de azúcar serían un biomarcador de diabetes) y el Dr Massi Bianchi de Ulysses nos enseñó datos muy prometedores de poder medir plasticidad neuronal en un análisis de sangre, si nos fijamos en dos marcadores en concreto. Otro tipo de biomarcador son los de técnicas de imagen cerebral, y el Dr Eric Marsh de Filadelfia nos enseñaba también datos de cómo hay algunas señales del electroencefalograma que correlacionan con la severidad de la enfermedad en CDD. Son dos tipos de marcadores que muy posiblemente se usarán en ensayos clínicos.
Luego el Dr Marsh nos compartió los resultados interinos de un estudio americano para desarrollar nuevas escalas clínicas para CDD. Ya han completado la primera parte y ahora las están refinando y confirmando, y tiene muy buena pinta. Y quiero destacar un detalle: este es un estudio observacional con más de 100 participantes, y 100 es justamente el número de pacientes de un ensayo de Fase 3 en CDD, así enhorabuena a la comunidad de pacientes por movilizarse de tal manera para hacer este estudio y los de los biomarcadores sean posibles.
Y por último el Dr Xavier Liogier, de la Loulou Foundation nos actualizó sobre el estudio CANDID, que es parte de un consorcio con varias empresas que están desarrollando tratamientos para CDD: El estudio CANDID busca validar en CDD escalas que ya se han usado en ensayos para otras enfermedades. Hace un año, Xavier nos contaba que el primer paciente ya había entrado al estudio. Ahora 12 meses después nos decía que ya tienen más de 100 visitas programadas con lo que cierran reclutamiento, y nos enseñó ya muchos de los datos que tienen viendo cómo estas escalas se aplican a CDD. El gráfico de reclutamiento al final parecía exponencial, y de nuevo muestra la tremenda movilización de la comunidad de pacientes para hacer este estudio posible.
El Dr Billy Dunn, que era el director de neurología de la FDA y ahora es Asesor Senior de la Fundación Loulou, fue moderador de esta sesión y alabó el espíritu de colaboración de los participantes por enseñarnos datos mientras los estudios están aún en curso, y destacó también el valor de estar construyendo “un menú del que elegir” para hacer posible diseñar ensayos complejos en CDD.

Proyecto de biomarcadores de Ulysses y curva de reclutamiento exponencial de CANDID
5. HACER UNA TERAPIA GÉNICA ES COMO CONSTRUIR UNA CASA
Hace un año, en el Foro 2022, pensábamos que en este Foro tendríamos la noticia de que una de las terapias génica había obtenido permiso para empezar ensayos. Pero todavía no hemos llegado a esa noticia.
Y hablando con algunos de los padres en el Foro sobre cómo explicar a gente que no es científica las implicaciones de que haya retrasos en este tipo de proyecto complejo, lo mejor que encontramos es el ejemplo construir una casa que compras sobre el plano. Si no lo habéis vivido directamente seguro que conocéis a alguien a quien le ha pasado: firmas sobre plano, el constructor te dice el plazo en el que te podrán dar las llaves, y al final raramente es en esa fecha. Y es que hay demasiadas etapas que pueden añadir retrasos, pero eso no quiere decir que un retraso indique que hay un problema con la casa o que no te la llegarán a entregar. Puede que hacer los cimientos se retrase por un invierno largo, el aislamiento haya que retocarlo, la estructura exterior de la casa nos de alguna sorpresa… añade a eso todas las instalaciones, los acabados exteriores y los interiores… hay demasiados pasos que pueden alterar esas predicciones de fechas.
Hacer una terapia génica es como construir una casa. Los expertos en biotecnología saben cómo hacer estas terapias, ya lo han hecho antes, pero aún así tienen que hacer la tuya desde el principio y hay muchos pasos complejos incluidas las inspecciones (igual que en una casa!) que acaban moviendo los plazos pero que son parte normal de hacer una terapia génica.
Lo que sabemos hoy es que tanto la terapia génica de Ultragenyx como la que está haciendo la Fundación Loulou con la Universidad de Pensilvania ya han hablado con las agencias reguladoras en 2023, y que ambas esperan obtener la aprobación para ensayos en 2024. Así que el proceso ha sido bueno, aunque no tan rápido como queríamos. Y Sharyl de Ultragenyx fue muy clara en este Foro: no se han encontrado ninguna señal adversa de seguridad con la terapia génica de CDD en ninguno de sus estudios con animales realizados hasta la fecha, incluyendo los estudios de seguridad en primates. Es simplemente un caso de tener que hace run poso más de trabajo para pasar la inspección de la casa.
En resumen al final del año pasado nos dijeron “creo que te daremos las llaves de la casa el año que viene” y no ha sido así, pero no hay ningún problema con la casa. Es que es de verdad muy complicado atinar con las predicciones en proyectos tan complejos.
Y en el Foro 2023 repasamos también los progresos en desarrollar terapias génicas con modalidades nuevas, de las que no hay aún precedentes aprobados para otras enfermedades. En concreto vimos un update del Dr Kyle Fink, que está desarrollando una terapia génica basada en CRISPR para abrir la segunda copia del gen CDKL5 en niñas (la del segundo cromosoma X), y de Kelcee Everette que trabaja en el laboratorio del Dr David Liu y que también está desarrollando una terapia génica basada en CRISPR, en este caso para arreglar la letra mutada o poner la letra que falte en el gen. En ambos casos se centran en aumentar la eficacia de corrección del gen, aumentar la selectividad (que no cambie genes que no tocan) y hacer los CRISPR más pequeños porque todavía no caben en los virus que se usan para terapia génica. Estos proyectos son como hacer casas con materiales nuevos, que no se han usado todavía nunca, y que de momento son demasiado grandes para la casa. Con lo que los plazos para su llegada a ensayos son impredecibles, pero el progreso es muy tangible. Y a mi me parece aún increíble que CDD sea de las primeras enfermedades a las que se están aplicando estas terapias génicas del futuro.

Varias terapias génicas en desarrollo
6. DOS SORPRESAS QUE HAN ABIERTO DOS PUERTAS
No todo está tardando más de lo que pensábamos. Hay cosas que están pasado más rápido de lo que podríamos imaginar, y vimos dos ejemplos notables de esto en el Foro.

Aprendiendo de la evolución
El Dr Ibo Galindo del Príncipe Felipe nos presentó un análisis evolutivo fascinante del gen CDKL5 desde la primera célula de la evolución y pasando por el árbol entero de la vida. Resulta que las plantas tuvieron en origen un gen CDKL, pero lo perdieron (¡tienen deficiencia en CDKL5!), que el gen CDKL5 original fue el 5, pero que luego se duplicó varias veces dando lugar a las 5 versiones. Y también aprendimos que mientras que las cinco formas CDKL1-5 tienen funciones importantes de regulación del esqueleto celular (el citoesqueleto), el gen CDKL5 parece haber adquirido funciones nuevas en vertebrados. Había dos conclusiones importantes: (1) es posible que haya redundancia funcional entre alguno de estos genes, y (2) CDKL5 tiene una cola especial en vertebrados que indica que tiene una función que no es de regular citoesqueleto.
Y estas conclusiones nos pusieron un marco de fondo perfecto, para dos proyectos que vienen del laboratorio de la Dra Sila Ultanir del Crick Institute y que os cuento más abajo. El laboratio de Sila se llevó el premio al Laboratorio del Año en el Foro 2023, pero a mi me gusta llamarlo el laboratorio de la década por el impacto que tienen en la investigación en CDD.
1. Redundancia funcional. Encontramos el parálogo. Encontramos avenida terapéutica.
En biología, un gen paralógo es el que nace por duplicación de otro, y ahora se parecen tanto que posiblemente compartan algunas funciones. Esto es lo que pasó en la atrofia muscular espinal, esa enfermedad neuromuscular letal en bebés que los científicos solucionaron porque encontraron un gen SMN2 (¡un parálogo!) que podía compensar por la falta de SMN1.
El laboratorio de Sila se dio cuenta de que una de las proteínas de citoesqueleto más famosas controladas por CDKL5, que se llama EB2, no estaba controlada solo por CDKL5 en neuronas, había otra kinasa que también la regulaba. ¡Y buscando quien era encontraron que era CDKL2! Y ahora están buscando la posibilidad de aumentar la expresión de CDKL2 en CDD para compensar por la falta de CDKL5. Y hay una posibilidad de hacerlo con ASOs. Una de las ponencias invitadas fue por el Dr Stanley Crooke, que es el fundador de la empresa Ionis (la que hizo el primer oligonucleótido antisentido o ASO para tratar la atrofia muscular espinal), y que luego a fundado una ONG llamada N-Lorem para hacer ASOs específicamente para gente que tiene enfermedades genéticas “nano-raras”. Y dijo en su charla que “CDKL2 tiene los elementos arquitecturales genéticos necesarios para hacerle un buen candidate a aumentar su expresión usando ASOs”. Y se eso sabe mucho este hombre.
Ha sido muy inesperado enterarnos de que quizás podamos seguir los pasos de la atrofia muscular espinal al encontrar un segundo gen que pueda compensar con el primero (al menos en neuronas, porque parece que en otros órganos son otros CDKL los que compensan). Ha sido un descubrimiento y comienzo de un proyecto terapéutico que nos ha llegado super-rápido.
2. Predicción que tiene dianas que no son de citoesqueleto. Encontramos la diana. Encontramos avenida terapéutica.
La Dra Marisol Sampedro, que trabaja con Sila, nos presentó el año pasado su descubrimiento de que CDD podría ser parcialmente una canalopatía porque encontró que el canal de calcio Cav2.3 era una diana clave de CDKL5. Cuando falta CDKL5, el canal Cav2.3 no está fosforilado (el interruptor que le pone CDKL) y se queda demasiado abierto, y la neurona se queda demasiado activa. Así que lo que necesitaríamos son inhibidores de este canal, y esto lo repasamos en el resumen del año pasado donde lo llamé un descubrimiento muy importante.
Pues un año después teníamos en el Foro una empresa nueva llamada Lario Therapeutics contándonos que están desarrollando fármacos inhibidores de Cav2.3 para CDD y para otro síndrome causado por mutaciones de ganancia de función en ese gen (también tienen Cav2.3 con demasiada actividad). El director científico de Lario nos explicó que están terminando la optimización química de los compuestos y que pasarán a los estudios de seguridad en animales previos a ensayos clínicos. Estamos hablando de una medicina de precisión para CDD y esto de verdad que nos ha llegado mucho más rápido de lo que podríamos haber imaginado.
RESUMEN: MOMENTUM
Este año aprendimos que la primera terapia génica está tardando más de lo que pensábamos, pero muchas otras cosas están pasando más rápido que lo que hubiéramos pensado, como esas dos nuevas oportunidades terapéuticas (CDKL2 y Cav2.3) que eran inimaginables hace tan solo dos años. También vimos por fin los frutos de varios esfuerzos de varios años de duración, como los dos estudios observacionales que han reclutado más pacientes que un ensayo de Fase 3, o tener por fin modelos de epilepsia in vitro y en ratas que permiten el tipo de experimento que necesitan las empresas farmacéuticas. Así que la impresión que yo me llevo es de que la velocidad de progreso se está acelerando, un poco como este gráfico de reclutamiento del estudio CANDID, y que hemos llegado a un momentum importante.
Y esa es justamente la palabra que Stan Crooke usó cuando se subió al podio y nos dijo “el momentum que tenéis ahora mismo en investigación en CDD es palpable”. Por eso creo que es buena palabra para capturar lo que vimos este año (aunque no la usemos mucho en español, es una especie de inercia inparable).
Y ese momentum se apoya sobre dos pilares que también se mencionaron mucho durante este Foro: la determinación y el espíritu de colaboración.

La voz de las familias en el Foro 2023
Así que os dejo con las frases de tres de los padres/madres que hablaron en el Foro 2023 y que capturan estos mensajes:
Cristina, de la Asociación de Afectados CDKL5, que nos recordó que “las personas con CDD son increíbles y se merecen lo mejor”
Majid, de la Fundación Loulou, que reconocía el camino que queda por recorrer explicando que “hemos avanzado más rápido y más lejos de que lo hubiera imaginado, gracias a todos vosotros. Pero al mismo tiempo, nunca es tan rápido ni tan lejos como querríamos las familias” y pedía a la audiencia que se volvieran a comprometer a curar la enfermedad con aún más pasión, más motivación y más determinación
Y por último Heike, en representación de la Alianza Internacional, nos hablaba de la personalidad de su hija, de los esfuerzos que los diferentes miembros de la Alianza están llevando a cabo, y cómo “estamos en esto juntos”.
Ya se que digo lo mismo todos los años, pero el de este año fue el mejor Foro que hemos tenido.
Espero que os haya gustado el resumen. Ya me diréis lo que os parece en los comentarios.
Ana Mingorance, PhD
Nota: este texto captura mis impresiones de las presentaciones del Foro que más me interesaron, no es un texto oficial del congreso emitido por la Fundación Loulou. Escribo estos resúmenes para los padres de personas con CDD, así que a veces me tomo ciertas licencias a la hora de explicar las partes mas técnicas.

MAIN LESSONS FROM THE 2023 CDKL5 FORUM
For the past nine years the Loulou Foundation hosts an annual meeting where scientists and drug developers working on CDKL5 deficiency, together with representatives from patient organizations, meet to discuss the latest advances.
Here are the main news and take-home messages from the 2023 CDKL5 Forum that took place in November 6-7 2023

2023 CDKL5 Forum
For the past nine years, the Loulou Foundation has hosted an annual meeting, the CDKL5 Forum, where scientists and drug developers working on CDKL5 Deficiency Disorder (CDD), together with representatives from patient organizations, meet to discuss the latest developments in the field and to advance towards treatments and cures. You can find summaries from the past few meetings here: 2018, 2019, 2020, 2021 and 2022.
The 2023 CDKL5 Forum edition took place November 6-7 in London, UK, and will return to Boston next year for the 2024 edition. Each year I say that this was the best Forum so far, but it is true. I wrote 47 pages with notes. There was so much good information and so many important moments! I will try to summarize the main take-home messages from this year’s Forum including the scientific pre-forum meeting. This won’t cover all the presentations, but rather focus on the main themes that we saw and the main progresses – which are many.
1. USEFUL NEW INSIGHTS INTO CDD
The Forum always opens and closes with the words from the patient community. This year, Cristina, from CDKL5 Spain, opened the conference with some powerful words. She explained that when they received their daughter’s diagnosis, they also learnt that their daughter “would live a life somewhere between uncertain and horrible”.
That’s why for her, research means hope.
And every year we keep learning new things about CDD that have important implications to understanding the disease and thinking about treatments. Here are 3 of those, and check out also point #6 in this summary.
Dr Lauren Orefice from MGH and Harvard showed us that food texture aversion in CDD happens because of sensory changes, rather than behavioral reasons. Mice with CDD have sensory neurons that are hyper-reactive to rough texture, so no wonder they don’t like it. This is probably the same for people with CDD who might have difficulties with specific food textures.
Several research projects keep finding functional changes in brains with CDD, rather than structural changes (Dr Tanaka, Dr Higley, Dr Fagiolini and others). This means that neurons are in the right place, and overall the brain is all well shaped and connected, but some neurons are firing together more than they should while others are not firing together as much as they should. To me, this is promising for treatments because normalizing neuronal activity can lead to plastic changes in this type of neuronal functional connectivity. What would be much harder is to try to correct having neurons in the wrong place.
We had a wonderful lecture by Sir Adrian Bird, who became famous after showing that Rett syndrome could be reversible in mice. He explained to us how in Rett syndrome too little or too much MECP2 are both bad options, so the gene therapies in development for Rett syndrome (too little MECP2) all incorporate control mechanisms to prevent running into overexpression MECP2. A big surprise from this Forum was Dr Sharyl Fyffe-Maricich from Ultragenyx confirming with data in mice and primates what we had suspected for several years: the body puts a limit to how much you can express CDKL5 and it will not let you get into overexpression (too much) of CDKL5. The neurons eat all of the excess away, no matter how much gene therapy you give them. This is very encouraging, because it makes gene therapies for CDKL5 easier since we don’t need to add that “break”.

Food texture aversion and learning from the Rett syndrome field
2. SOLVING KEY PROBLEMS TO ADVANCE DIAGNOSIS AND RESEARCH
The 2023 Forum showed even more collaborative reagent and model generation than previous years, and in particular I would like to highlight advances around diagnosis and seizure models.
Genetic testing is not always easy to interpret. For example, if your child has a mutation in the CDKL5 gene that breaks the gene, then it is easy to interpret in a genetic test. But what happens when the mutation changes a letter by another one? This one is hard, and we end up with VUS (variant of unknown significance) and an uncertain diagnosis. A team from the Telethon Institute of Genetics and Medicine (TIGEM) is making in the lab all of the possible letter changes in the CDKL5 kinase domain and will check all of their functions in cells and then put the results in a database. That way, genetic labs will always have an answer for any letter change that they find. No more VUS for CDKL5, what a cool project.
And another big research tool progress from this year was that lots of seizure-relevant models are finally available! We had Dr Liz Buttermore show us a beautiful plate assay with cells from patients with a very nice hyperexcitability signal in patient-derived neurons. As a former pharma scientist, I know this is the type of assay that I would like to use. If you remember from other years, Dr Muotri from UCSD had a great in vitro CDD model using organoids from brain cortex. This year Dr Rebeca Blanch from his lab showed us that they are now making thalamo-cortical organoids, to try to better recreate one the main epilepsy circuits. Very cool! And Prof Peter Kind showed us how his lab has identified an epilepsy hotspot in CDD rat brains and they are able to stimulate it to induce seizures whenever we want. That’s such as great epilepsy model! This year we had incredible progress in this area.

VUS databases and CDD rat seizure model
3. “THE APPROVED 4”, AND TREATMENT PIPELINE TODAY
CDD had the first drug approved in the US (2022) and Europe (2023).
To show you just how special and how hard it is to have a drug approved for a rare epilepsy syndrome, I will repeat here what I said in my talk during the treatment pipeline session: there are more than 300 epileptic syndromes, and there are only 4 epilepsy syndromes that have any drug approved. These are Tuberous Sclerosis Complex (TSC), Lennox-Gastaut Syndrome (LGS), Dravet syndrome, and CDD. That’s it, just 4.
A pharma company recently started referring to these as “the approved 4” because it is so exceptional for an epilepsy syndrome to have a drug that has made it all the way to the finish line. For companies looking to develop treatments, these approved 4 are going to be easier and therefore more attractive than the others, because there is already a precedent and therefore many answers to how to get a drug to approval for them. So the value of ganaxolone goes beyond the value of the medicine.
We have gone from zero to 8 clinical trials in 8 years and have become part of the approved 4. This is a very strong beginning.
Now the question that remains is “what’s next”. At the Forum we had several presentations for the therapies that come next and many of the efforts to support them, which I summarize here and in the next three sections.
Marinus Therapeutics announced at the Forum that they have now signed a Global Access Program to help get ganaxolone to people with CDD in countries where it is not commercially available (or in process of becoming available, like is the case for Europe). Their website has a contact email for doctors to reach out to learn more about access. They also showed nice data of sustained seizure efficacy with ganaxolone over 2 full years.
UCB Pharma was represented by two speakers, including their CMO, who explained that UCB spends 30% of their revenue in R&D – this is a lot for the industry. And as you know some of that went to acquire Zogenix and their drug fenfluramine, which is now in Phase 3 trials for CDD. The trial has a website with an easy to remember name: cddstudy.com and you can see there all the countries where the trial is available in Asia, Europe and North America.
We also reviewed several drugs in development for other epilepsies or other neurodevelopmental disorders and that could potentially join the CDD pipeline in the coming years. These included other GABA drugs, KCC2 activators, Cav2.3 inhibitors (which we review in section 6) and some treatments that target the gut microbiome and that I find very interesting.
The conclusion for the session on the current CDD pipeline was that ganaxolone is just the beginning and that are multiple options coming behind it.

CDD therapeutic pipeline presentations
4. CLINICAL TRIAL READINESS: HOW THE PATIENT COMMUNITY IS MAKING THE FUTURE THERAPIES POSSIBLE
After the Forum we got emails from pharma and biotech industry professionals that attended the Forum and they all mentioned this session as very impactful. As one literally wrote, “this will attract more industry participation”.
This session was about biomarkers and outcome measures, which is “what to measure in trials that will look at more than seizures”. This is particularly critical for the future gene therapy trials, and the last 12 months have seen tremendous progress.
Biomarkers are way to see changes in the brain without directly taking a biopsy. One type of biomarkers are the ones we can measure in blood (glucose levels are a biomarker for diabetes), and Dr Massi Bianchi from Ulysses showed us very promising early data for measuring neuronal plasticity in a blood test by looking at two key markers. Another type of biomarker is using brain imaging, and Dr Eric Marsh from CHOP showed us data from a large collaboration where they can correlate EEG signals with disease severity in CDD patients. Both of these types of biomarker are very likely to be used in future clinical trials.
Dr Marsh also updated us on the development of novel clinical scales to measure CDD symptoms beyond seizures run by the ICCRN network. They have completed a first part and they are now tweaking and confirming the new scales, which are looking really promising. And one important bit to highlight: this is a study with over 100 patients, and 100 patients is the size of a Phase 3 trial in CDD so kudos to the patient community for mobilizing to make this work and the biomarker studies possible.
And then Dr Xavier Liogier, from the Loulou Foundation, updated us on the progress around the CANDID study which is run in collaboration with a consortium of companies developing CDD treatments. This study evaluates well-known clinical scales that have already been used in clinical trials but for other diseases, to see how they may apply to CDD. Last year Xavier told us about the first patient having been enrolled. Twelve months later he told us that we will get more than 100 participants by the end of this year and showed us a lot of data for how these scales are able to capture different CDD symptoms. The recruitment graph looked exponential, again showing the tremendous mobilization of the patient community to make the CANDID study possible.
Dr Billy Dunn, former Director of the Office of Neuroscience at FDA and current Senior Advisor to the Loulou Foundation, moderated this session and praised all presenters for the spirit of collaboration in sharing all these data while still in progress, and highlighted the value of building all these biomarkers and scales as a “menu to choose from” to enable complex trials in CDD.

Ulysses biomarker project and exponential CANDID recruitment curve
5. MAKING A GENE THERAPY IS LIKE BUILDING A HOUSE
One year ago, at the 2022 Forum, we through that by the end of 2023 we would have the announcement that one of the gene therapies for CDD had received green light for a trial. We didn’t get this news.
And discussing with some parents at the Forum about the best way to explain to non-scientists the implications of delays in very complex process projects, we came up with the idea of how this is very much like building a house from scratch. You approve the initial design, and the constructor gives you a timeline for when you will get the keys. Then it rarely ever happens as planned. And that is because there are too many steps that can end up taking a bit longer, but this is not a sign that there is a fundamental problem with the house or that you will never get the keys. It may be that pouring the foundation is delayed due to a long winter, the insulation had to be redone, building the skeleton of the house can also unlock some surprise… add to that installations, exterior finishes, then all of the interior work… there are just many steps that impact timeline predictions.
Making a gene therapy is not unlike building a house. The biotech experts know how to make gene therapies, but they still have to adjust each step to our particular gene therapies and there are so many complex steps including regulatory reviews (like the different house inspections!) that timelines end up shifting but that is just a normal part of making a gene therapy.
What we know today is that the gene therapy from Ultragenyx and the one that the Loulou Foundation is developing with UPenn have indeed both already been presented to regulators in 2023, and are both now hoping to get clinical trial approval in 2024. So the progress has been good, just not as fast as we all hoped for. And Sharyl from Ultragenyx was very clear in her presentation this year: they have not found any adverse safety signals with the CDD gene therapy to date in any of their studies in animals including primates. This is a case of having to complete some more extra work to pass the house inspection.
Bottomline: at the end of last year we were told “I think I can give you the keys to the house by the end of next year” and that didn’t happen yet, but there is no problem with the house. It is just really hard to predict the exact timelines in very complex projects.
At the 2023 Forum we also reviewed the progress using new gene therapy modalities that haven’t been yet approved for any human disease so there are no precedents. In particular we had an update from Dr Kyle Fink from UC Davis who has been developing a CRISPR-like approach to reactivate the second copy of CDKL5 gene in each cell in females (from the second X chromosome), and also from Kelcee Everette from David Liu’s lab who is also developing a CRISPR-like approach called prime editing to correct the wrong letter or inset a missing letter in CDKL5 mutated genes. In both cases, much of the work focuses on improving efficiency (more copies of the gene fixed), specificity (not mess up with other genes) and figuring out how to make the CRISPR tools smaller so that they can fit the viruses used in gene therapies. These projects are like building new houses with completely new materials that had never been used before, and that so far are too large to fit well into the house. So timelines are unknown, but progress is very tangible. And it is quite amazing to see that one of the first diseases that these new technologies are being applied to is precisely CDD.

Multiple gene therapies in development
6. TWO SUPRISES THAT HAVE OPENED TWO IMPORTANT NEW DOORS
Not everything is taking longer than we thought. Some things are happening faster than we thought. We saw two great examples of this at the Forum.

Learning from evolution
Dr Ibo Galindo from the CIPF presented a fascinating evolutionary analysis of CDKL5 from the first single cell that existed and throughout the entire tree of life. It turns out that plants had CDKL genes but lost them (they have CDD!), and the original CDKL gene was CDKL5 but then it got duplicated twice leading to 5 forms. And it turns out that while all CDKL1-5 forms appear to be important to control cell skeleton, CDKL5 seems to have acquired new functions in vertebrates. There were two important conclusions: (1) there is probably functional redundancy among CDKL proteins, and (2) CDKL5 has a unique tail in vertebrates that suggest a new function not related to cytoskeleton.
And those conclusions provide a beautiful context to two projects coming from the lab of Dr Sila Ultanir at the Crick Institute that I summarize below. Sila’s lab was awarded the Lab of the Year Award by the Loulou Foundation in this last Forum, but I like to call them the “Lab of the Decade” because of their major ongoing contribution to the CDD field.
(1) Functional redundancy. Found the right paralog. Found a new therapeutic avenue.
A paralog is a gene that was born from a duplication of another gene, so now the two copies are so similar that they might have overlapping functions. This is what happened in SMA, the neuromuscular disease that is lethal in babies born without any functional SMN1 gene, that is now treatable because scientists found a way to use SMN2 (a paralog!) to compensate for the loss of SMN1.
Sila’s lab found that the most famous cytoskeleton target that we know for CDKL5, called EB2, wasn’t only controlled by CDKL5 in neurons, there seemed to be another kinase that could do the same job. And it turned out to be CDKL2! So now she is looking into the possibility of upregulating CDKL2 to compensate for the loss of CDKL5. And there is a potential to achieve this using an ASO approach. At the Forum we had a wonderful lecture by Dr Stanley Crooke, who founded and led Ionis (the company that made the first ASO treatment for SMA) and later created a non-profit organization called N-Lorem to make ASOs specifically for people with nano-rare diseases. And he explained that “CDKL2 has nice architectural features to make it suitable for overexpression” using ASOs. And he knows about this!
It has been quite unexpected to find out that we might get to follow the steps of SMA by finding a second gene that could do much of the work of the first gene (at least in neurons, because looks like in other organs it could be another CDKL). And this happened super-fast.
(2) Predicted to have targets beyond cytoskeleton. Found the right target. Found a new therapeutic avenue.
Dr Marisol Sampedro, working in Sila’s lab, had made the discovery last year that CDD could be partly a channelopathy because she found a new target for CDKL5 that is an ion channel. The channel is called Cav2.3, and when it is not phosphorylated by CDKL5 (as would happen in the deficiency) the channel stays too open, and the neurons stay too active. So what we would need is inhibitors. You can read about this in last year’s Forum recap (point 4, where I called this discovery a breakthrough).
One year later we had a new company standing on stage, called Lario Therapeutics, telling us that they are developing Cav2.3 inhibitors for CDD and for a disease caused by gain-of-function mutations in that gene (so they also have too much activity). Lario’s chief scientist explained that they are close to finishing the chemistry optimization and to start running safety experiments to advance their new drug towards clinical trials. This is a precision medicine approach to treat CDD and this seriously happened super-fast.
SUMMARY: MOMENTUM
We learnt this year that the first gene therapy is taking a bit longer that we thought, but many other things are happening faster than we thought, including having new therapeutic avenues (CDKL2 and Cav2.3) that were unimaginable for us barely 2 years ago. We also had several multi-year-long efforts all give us tangible results finally this year, like the two observational studies that have recruited as many patients as Phase 3 trials, or finally having beautiful in vitro and in vivo epilepsy CDD models of the type that pharma companies need. It feels to me that the pace of progress is accelerating, almost like that recruitment timeline for the CANDID study, and we have reached a powerful momentum.
That is exactly the same word that Stan Crooke chose when he stood on stage, and told us how “you can feel the momentum behind the science” in the CDD space. And I think that is a good word to capture what we saw this year.
And this momentum builds on two pillars that were also mentioned often during the 2023 Forum: determination and the spirit of collaboration.

Voice of the patient at the 2023 CDKL5 Forum
So I will leave you with quotes from three of the parents that spoke at the meeting that capture some these messages:
Cristina, from CDKL5 Spain, reminding us that “People with CDD are amazing and they deserve the best”.
Majid, from the Loulou Foundation, acknowledging the road to come with “We have moved faster and further than I expected thanks to all of you. And at the same time, that’s never as fast or as far as hoped for by the families”. He then called upon the audience to recommit with even more passion, more drive and more determination.
And Heike, representing the CDKL5 Alliance, who told us about his daughter and her personality, the efforts that all countries in the alliance and doing at the local and international level, and how “together we are stronger”.
I know I say this every year, but this was the best CDKL5 Forum so far.
I hope you enjoyed this summary. Please let me know your thoughts in the comments.
Ana Mingorance, PhD
Disclaimer: This is my own summary and key learnings, and not an official text about the Forum by the Loulou Foundation. I write these texts with the parents of people with CDD in mind, so excuse also my lack of technical accuracy in parts.

Dónde está la mutación de tu hijo en CDKL5, y qué nos dicen los gemelos
Una conversación que suele salir mucho en reuniones de familias CDKL5 es la posición de la mutación de sus hijos en el gen, por si eso nos ayuda a saber la severidad de la enfermedad cuando crezcan. Os resumo abajo un listado de publicaciones y sus conclusiones.
Una conversación que suele salir mucho en reuniones de familias CDKL5 es la posición de la mutación de sus hijos en el gen, por si eso nos ayuda a saber la severidad de la enfermedad cuando crezcan.
Yo sé que hubo estudios que decían que las mutaciones hacia el principio del gen son peores, y que si son mutaciones hacia el final “como tienes casi toda la proteína” dan lugar a cuadros clínicos más leves.
Pero también sé que hoy en día ya no se cree en esa correlación. Pero para estar seguros he mirado la literatura para ver si aún creemos que hay correlación entre mutación y fenotipo, o si no. Os resumo abajo un listado de publicaciones y sus conclusiones.
Nota para el lector no experto:
Genotipo es el tipo de mutación
Fenotipo es el cuadro clínico, los síntomas)
2004 - Mutations of CDKL5 Cause a Severe Neurodevelopmental Disorder with Infantile Spasms and Mental Retardation
Artículo original que identifica CDKL5 como la causa del síndrome. Describen una familia que tuvo 5 hijos, y eso incluye dos sin problemas, un varón que se les murió con 16 años con discapacidad intelectual several y epilepsia, y gemelas idénticas ambas afectadas. Eso es lo que llevó a los médicos a buscar la causa genética más que probable del problema de los tres hermanos, y encontraron que era CDKL5. Ahora eso eso, una de las gemelas idénticas tenía un cuadro clínico de “síndrome de Rett” (que resultó ser CDD) mientras que la otra, gemela idéntica, solo tenía trastorno de espectro autista. Basado en las descripción, con la misma mutación, el hermano tenía un fenotipo muy severo, una de las hermanas un CDD promedio, y la otra muy altamente funcional.
La familia 2 tuvo también dos casos, ambas hijos d ella misma mujer aunque de diferentes padres. Una considerada Rett atípico porque siempre estuvo muy afectada, y otra Rett clásico porque empezó mejor y a partir de los 3 años perdió uso de manos y dejó de adquirir más capacidades motoras. Misma mutación.
Con estas dos familias, sobretodo con las gemelas idénticas, está claro que sabiendo la mutación NO puedes saber si será más o menos afectado.
2012 - Recurrent mutations in the CDKL5 gene: genotype-phenotype relationships
Miran a niños que llevan la misma mutación en CDKL5 y si se desarrollan de forma igual. Ven que hay una mutación repetida (una missense, de cambio de letra) que produce casos más ligeros, pero todas las otras repetidas más severos. Y de esas las hay hacia el principio y hacia el final.
2012 - What We Know and Would Like to Know about CDKL5 and Its Involvement in Epileptic Encephalopathy
Explican “hasta ahora no se ha visto correlación clara de genotipo-fenotipo en casos de mutaciones en CDKL5. Algunas publicaciones sugieren que mutaciones hacia el final del gen dan lugar a cuadros clínicos menos severos, pero otros argumentan que la naturaleza de la mutación no tiene impacto en el espectro clínico. Por ejemplo el estudio de Weaving y colabores que describe dos gemelas idénticas con un fenotipo muy opuesto, una con un fenotipo que concuerda con Rett y la otra con espectro autista pero de discapacidad intelectual leve. Como se les miró la inactivdación del cromosoma X y ambas lo tenían normal, se piensa que su diferencia de fenotipo se debe a genes modificadores o factores ambientales"
2014 - Mutations in the C-terminus of CDKL5: proceed with caution
Algunos casos de mutaciones cerca del final del gen se ve que estaban mal interpretados, que se los había considerado patogénicos pero no eran porque eran heredados de los padres (todos llevamos muchas letras cambiadas en nuestro ADN, la inmensa mayoría de las veces no son problemáticas). Avisan de que las mutaciones en los exones 19’21 podrían ser falsa interpretación y tener cuidado con esos resultados genéticos.
Me pregunto si este descubrimiento también se juntó con la creencia de que mutaciones al final son menos malas. Estas no eran mutaciones, eran variantes que tenían esas familias igual que tenemos todos variantes de muchos genes.
2015 - Two Siblings With a CDKL5 Mutation: Genotype and Phenotype Evaluation
Dos hermanas, con la misma mutación (a principio del gen). La primera tiene retraso de desarrollo severo, hipotonía muy alta desde que nació, con problemas para comer con lo que lleva gastrostomia, y crisis refractarias. La segunda no tiene síntomas hasta 3 meses, cuando le viene la epilepsia y los síntomas. Hasta entonces ni hipotonía ni nada. Ya tras empezar las crisis si que deteriora, y consideran que tiene encefalopatía epiléptica. Al final la pequeña está más afectada que la mayor. Piensan que quizás sea por lo de la inactivación del cromosoma X, que quizás la pequeña tenga más neuronas con el gen mutado. Lo que importa es que con la misma mutación desarrollan distinto, y que la que empezó menos severa al final tenía la peor enfermedad. Con lo que no se puede saber.
2020 - CDKL5 Deficiency Disorder: Relationship between genotype, epilepsy, cortical visual impairment and development.
Este es un artículo de de los mayores especialistas de CDD en estados unidos. Explican que “los análisis de correlación de genotipo-fenotipo han sido difíciles en CDD porque no había suficientes pacientes y porque teníamos pocas mutaciones repetidas” y por eso juntan 92 pacientes, para ver si algo correlaciona, y concluyen que “ningún parámetro de la enfermedad correlacionaba con el genotipo”
2021 - Exploring genotype-phenotype relationships in the CDKL5 deficiency disorder using an international dataset
Usan una base de datos de 285 casos para ver si hay correlación entre el tipo de mutación y el desarrollo. La severidad de la enfermedad la encuentran igual en todos los tipos, si acaso si la mutación es trúncate hacia el final del gen (después del aminoácido 781) podrían estar un poco mejor cuando se les mira como grupo, o sea sabiendo que siempre hay diferencias individuales.
Describen algunas variantes individuales que están peor, pero a mi personalmente no me gusta interpretar así estos resultados porque una persona es combinación de muchos factores no solo la mutación en concreto con lo que el hecho de que el niño más afectado tenga la mutación X, no quiere decir que un segundo niño la vaya a tener igual. Eso es lo que vemos con hermanos, que salen distintos aún llevando ambos la misma mutación.
2022 - Factors influencing the attainment of major motor milestones in CDKL5 deficiency disorder
Usan una base de datos de 350 casos (la misma que en 2020 pero con más casos) para ver qué factores predicen el mejor desarrollo de las personas con CDD, sobretodo los que consiguen sentarse solos. Si que dice que si la mutación es trúncate hacia el final del gen (después del aminoácido 781), o ser niña en vez de niño, tienen mayor probabilidad de llegar a sentarse solos. A los que la primera crisis les empieza más tarde también tienen mayor posibilidad de llegar a sentarse solos. Pero cuando miramos los datos para ver cuanto más de probabilidad tienen de llegar a sentarse: un bebé que tiene una mutación sin sentido (de parada prematura) al final del gen tiene una probabilidad de un 70% de que se pueda sentar solo cuando tenga 5 años, y uno con una mutación sin sentido (de parada prematura) al principio del gen tiene una probabilidad de un 55% de que se pueda sentar solo cuando tenga 5 años. O sea no quiere decir que mutación temprana implica caso severo, o que mutación tardía (en el gen) anticipe que el niño o niña será de los que caminan y hablan.
ULTIMO:
Por último, el blog de genética de epilepsias resume que:
“Análisis de genotipo-fenotipo se han hecho pero no han conseguido ver ninguna correlación consistente. Y la observación de que gemelas idénticas tienen diferentes fenotipos enfatiza que juegan mucho papel otros factores ya sean genéticos, epigenéticos o ambientales"
Y esta es la conclusión con la que yo estoy más de acuerdo. Que no tenemos datos para pensar qué saber la mutación del niño anticipa su desarrollo. Al contrario, tenemos datos para saber que con la misma mutación pueden llegar a tener desarrollo muy variable.
Ana Mingorance, PhD

IEC2023 Dublin
The International Epilepsy Congress (IEC) is one of the largest epilepsy meetings attracting clinicians, researchers and the pharmaceutical industry. In 2023 it took place in Dublin, in early September. I review the progresses towards treating the cause of the rare epilepsies / DEEs
The International League Against Epilepsy (ILAE) hosts every two years an international epilepsy congress (IEC) that just took place in Dublin. The main conference in the field is still the American Epilepsy Society meeting that takes place every year in early December (check out my summary of the 2022 AES meeting) but the IEC is a close second.
This text is not meant to be an exhaustive review of all what was discussed at the IEC. These are my highlights, as a rare epilepsy scientist who has been attending this type of conference for many years. Fun fact: did you know that in 2013 this conference was also held in Dublin? That was the year I decided to leave pharma to be able to work directly with rare disease patient groups. So this conference was very special for me, being back at the IEC in Dublin exactly 10 years later.
Here are the 9 messages that grabbed my eye during the IEC2023 conference, plus a bonus message. As usual, I focus on the development of treatments for rare epilepsy syndromes or DEEs.
1. Voice of the patient in the agenda

Amber Freed from SLC6A1 Connect
The IEC organizers did a great job at having many of their sessions open with the voice of the patient. And not just in the small rooms.
Amber Freed from SLC6A1 got to open the Presidential Symposium of the conference. The day before we heard from Gabi Conecker about SCN8A, and both supermums told the audience of clinicians about how the complexity of the DEEs mean that (1) we need coordinated multidisciplinary care, and (2) doctors need to listen to families. Listen not only because it is a nice human thing to do, but also because people with DEEs can do strange things that is not clear what they are (seizures? stereotopies? movement disorder?) so doctors really need to listen to families to understand what’s happening.
These are not new messages, but it is a new stage: the BIG stage. And such change is important to normalize the message that families are central to how we take care of people with DEEs, and that for that to happen we need to regard them as peers, including sharing stage. No more sitting at the back of the room listening to clinicians.
2. Comorbidity doesn’t mean comorbidity
Dr Scott Demarest from Colorado explained this very well. He said that:
We always talk about “comorbidities” because we are epileptologists, so everything that is not epilepsy is a comorbidity. But really these are also defining features of a syndrome, not necessarily set off to the side.
Dr Antonio Gil-Nagel from Madrid also explained later in the conference that only 3% of patients with DEEs have only seizures, so almost everyone has many more things going on: intellectual disability, developmental delay, endocrine problems, motor problems, language problems (the % of non-verbal people with DEEs is high!). He said that in his practice, families often report behavioral problems as their top priority, with aggressivity and impulsivity causing much stress to families.
I’ve also heard this a lot from families (Syngapians and PHD19 girls, I’m thinking of you here)
For me it was clear at the IEC2023 that this “comorbidity doesn’t mean comorbidity” message is sinking in, not only because we heard it from everyone (parents and doctors on stage) but also because there were several sessions dedicated to some of the non-seizure aspects of DEEs. And remember that this is an epilepsy conference! not a neurodevelopmental meeting, so this is really a big step towards helping look at these diseases more globally, which is an absolute requirement towards being able to manage them more globally.
3. Changing the therapeutic goal and how we talk about DEEs
Dr Elaine Wirrell from the Mayo Clinic explained the therapeutic goal for DEEs and how it is different from managing a patient who only has epilepsy. For epilepsy it usually is to try to get to seizure freedom with no (or manageable) side effects. For DEEs, which are so tough to treat, the goal of zero seizures would mean excessive sedation. Instead, the therapeutic goal should be:
“target the most problematic seizures, avoid therapies that might worsen seizures, avoid excessive polypharmacology, and manage the comorbidities to maximize the quality of life”
But that is now, when all we have is anti-seizure drugs and some anti-anxiety or anti-depressant medications, melatonin and others to sleep… all symptomatic! (and rarely very effective). The therapeutic goal will change once we have access to real disease-modifying therapies, which I talk about in some of the next sections.
Dr Elaine Wirrell also gave another very good talk where she reminded collages that words do matter, and to please stop using the term “catastrophic epilepsy”, in particular when explaining to parents what their child has been diagnosed with.
I have a friend (and Dravet dad) called Julian who would often start his presentations with exactly that word, “catastrophic”, as the single word in a slide. And would pause there. To let it sink into the bones of everyone in the audience. Catastrophic means that nothing can be done, it erases all hope.
And Dr Wirrell was fantastic at reminding us that catastrophic is not a medical term, and that:
“words matter! We CAN make a difference for people with developmental and epileptic encephalopathies and they ARE NOT catastrophic!”
She was a fantastic speaker.
4. What is disease modification?
Another of my favorite speakers was Dr Scott Demarest from Colorado. He spoke about how disease-modification “means we have to care about things other than epilepsy” and commented on some of the efforts in development to measure those other symptoms during clinical trials. As he explained, a clinical trial with a disease-modifying therapy (like those correcting a gene problem) has to be different from epilepsy clinical trials. This is not about counting seizures for 12 weeks, this is about counting a lot of new things for a lot longer than 12 weeks. For some of the syndromes we might not even be able to use seizure frequency as the primary endpoint (not enough or hard to count), but that’s ok, we are now seeing more assessments developed for many other disease aspects.

Dr Scott Demarest on disease-modifying trials for DEEs
Dr Demarest explained that disease-modifying therapies are treatments that target the fundamental basis of a condition, and that aim to improve the entirety of the phenotype, but do not necessarily mean “cure” in the sense of erasing the disease completely. But most DEE families that I know already call “cure” a treatment that targets the gene and improves several of the symptoms, so I think we agree on the goal: tackle the biology, and give the body a chance to do its best.
And I would like to take a second to pause here, and let you all realize how this conversation is an inflexion point in the field. This is the International League Against Epilepsy talking about disease-modification (!!), talking about cures, about fixing genes, about getting those treatments through clinical trials. All center stage.
As Prof Ingrid Scheffer from Melbourne said later in the conference, “if you came from the Stoke session, you know that gene therapies are here and now”.
Let that sink in.
It is not just the Stoke clinical trial. It is the whole field. It is a whole pipeline of treatments designed to target the genetic problem of several of the DEEs (many not presented at the IEC2023). This is an inflexion point in the field. Disease-modifying therapies are here and now.
5. Talking a lot about outcome measures
The imminent arrival of therapies that can do more than reducing seizures is changing how we think of clinical trials. Now we want to measure much more, and measuring more means having the scales or assessments to measure those things. That’s called clinical outcome measures.
Outcome measures are not a synonym with a comorbidity, they are scales to measure that comorbidity. It is sometimes confusing, also if we add the word endpoint which is the “goal” of moving that scale. Here is a simple example:
Imagine that you want to gain muscle mass to be able to get better at weightlifting:
The two “comorbidities/symptoms” to measure: weight and strength
Outcome measure 1: scale that can discriminate muscle and fat
Endpoint 1: two more Kg of muscle in X months
Outcome measure 2: measuring the maximum amount of weight you can lift for one repetition
Endpoint 2: increase the weight that you can lift for one repetition by X Kg
So as you can see we start getting into the technical aspects of clinical trials, like the specific assessments that we should use, which by the way should meet the requirements for being used for drug assessment, and also deciding what the goal change should be (the endpoint).
By the way, Clinical Outcomes Research is like a whole field of science! And here we have the clinicians who treat patients with DEEs rolling up their sleeves and getting fully fluent on the technicalities to develop outcome measures and endpoints that can help us run the new clinical trials. IEC2023 looked nothing like the 2013 edition, which was all about using approved anti-seizure drugs better. I tell you, this is an inflexion point in the field. And there is much more community involvement in the field, counting both clinicians and patients, than ever before.
This also opened up interesting debates: not all assessments are equal, so conclusions should NOT be also considered equally. Some of the companies with drugs approved to treat seizures in DEEs are using surveys or scales that capture the parent’s perception of how their child is doing to say that their drug can improve behavior or cognition. A doctor from the audience said very strongly (and rightfully) that you cannot claim that a drug can improve cognition if you don’t use psychological assessments to confirm that. So by all means you should listen to parents to understand the patient experience on a particular drug, but you need to go beyond that and involve neuropsychologists if you want to make certain claims of efficacy.
In the example above about weightlifting, it is not about whether you feel stronger, or your coach thinks that you are stronger, it is about whether we can verify that increase in muscle mass and performance.

Prominent stands with drugs for Dravet syndrome and LGS
6. A big part of the agenda is about DEEs…but mostly about three of them
DEEs were part of pretty much every session at the IEC2023, and often the only subject of entire sessions.
Much of this is because we are diagnosing many more of the syndromes, and learning about them, so at some point the discussion on seizure types (focal onset and generalized) takes as much space as the discussion about different syndromes.
But part of this is because pharma companies sponsor many of the sessions, and they of course want to talk about the diseases that they have drugs for. So with Epidyolex and Fintepla and Diacomit we got to hear a lot about Dravet syndrome, Lennox-Gastaut syndrome, and TSC. You could hear about these diseases in the exhibit area and also in all the sessions that companies sponsored.
And this is great as a field! But it was painfully narrow to focus so much on those few diseases. I understand that this is because a big focus of the conference is to learn about the medicines that are available and how to use them, and we now have several medicines for those syndromes. And also these are some of the most common syndromes so clinicians end up seeing many more of these patients than those with more rare DEEs. But I still believe the agenda skewed a bit too far.

Prof Ingrid Scheffer from Melbourne gave us a number that was truly eye opening: there are 925 monogenic epilepsy genes, and 825 of those explain about half of all DEE cases (others might not be genetic or a gene has not been found yet). That’s a lot!
Compare that to the focus on three syndromes.
I know that there were several sessions on genetics and I missed those (I read Dr Dennis Lal on Twitter to know what was said: @LalDennis). But those sessions focus largely on genetics as a way to DIAGNOSE, not as a way to TREAT. I think the only exception was the talk “Single gene approaches to precision medicine” precisely by Dennis Lal.
What would I like to see at a conference? I would like to see more discussions on DEE as a class, including all of the very rare genetic ones that we keep discovering year after year. Rare genetic DEEs have two things that make them different from other DEEs, so they need more space in this type of conference or we will not be prepared to treat them:
1) They are genetic, so we can think of ways to develop disease-modifying treatments for every single one. Isn’t this great? We might not know the best drug to treat a teenager with LGS but it is very straightforward to know what a child with a genetic DEE needs – the tricky part is to make that treatment. Some will be small molecule drugs to correct an ion channel activity (if that’s the cause of their DEE, like for SCN8A) or an ASO to change the protein expression levels (like for SYNGAP) or a gene therapy to put a gene back (like for CDKL5). Yes we will still help these patients with anti-seizure drugs, but we should talk more about all of the DEEs that have known genes and the different ways we could try to correct that problem and how we can turn those ideas into realities. If we do that, we will bring much more biotech (not just old pharma) to the table, and we really need that.
2) All genes produce a spectrum of phenotypes, and this impacts clinical trials. The clinical diagnoses for DEEs describe an electroclinical profile that is relatively homogeneous, that’s why clinicians have given it a name. But the genetic diagnoses of DEEs all produce a spectrum of symptoms and prognosis. That’s why you can have both Lennox-Gastaut syndrome as your epilepsy type, and CDKL5 deficiency disorder as your genetic diagnosis for example. So clinical trials for the majority of the DEEs, which are genetic diagnoses, are going to be a lot harder, and need more complex trials, relying more on non-seizure outcome measures. They cannot be left off the stage as we focus on Dravet syndrome and LGS and a few other clinical diagnoses (the ones that are good for epilepsy trials).
All that said, I think this is also about a field having been studied for longer. I liked how Dr Rima Nabbout from Paris acknowledged that we were talking a lot about Dravet syndrome at the conference, but also WHY it has become likely the most popular epilepsy syndrome at the meeting. She said:
“Dravet syndrome is emblematic because many years ago there were families that decided that they wanted to work hand in hand with the clinicians, with the pharma industry, and with the whole world”.
And she is 100% right. 12 years ago I started working with Dravet syndrome families to figure out how to make the disease better known to industry. There was only Diacomit and only in Europe. There were no plans for more trials, no companies interested. Dravet syndrome today, is many of the monogenetic DEEs in 5-10 years.
7. Adult neurologists step up
As we get better about diagnosing DEEs, and about taking care of these patients, we have more and more adults with DEEs. This contrasts with adult neurologists receiving a lot less training on these “childhood epilepsies”. And ironically, we are now running trials in patients under 18 years of age in these populations so some drugs get approved only for 18 and younger, or approved for more ages but without data in adults.
And to quote Dr Nabbout again:
“if children are not small adults, it is important to know that adults are not big children”
As patients grow, often the main concerns about their disease change. The parents of a small child with a DEE might worry mostly about seizures, but the parents of a 25 year old with a DEE are more concerned with aggressive behaviors and their son’s declining ability to walk independently, for example. It is important to also make sure that the usual multidisciplinary (and more coordinated) care of pediatric patients is also applied to adults after they transition to adult care, but this doesn’t happen so often. The International League has created a taskforce on transition (from pediatric to adult medical care), and it is led by Dr Nabbout. This is great.
8. Hot drug #1: XEN1101
This drug is not in development for any DEE, but looks great for focal-onset and generalized seizures (currently in Phase 3 trials for both) and is a good example for the type of small molecule drug that we could develop for different types of DEE. XEN1101 opens up an ion channel called Kv7, so that more potassium can flow through. There is one DEE caused by mutations in that very same potassium channel gene, and patients need exactly a drug that can open up the ion channel Kv7 because they only have half of the number that they need (KCNQ2 haploinsufficiency).
This is one of the most exciting drugs in development for epilepsy right now, and could be very useful for patients with KCNQ2-related epilepsy once it gets approved. For many of the DEEs caused by mutations in ion channels, small molecule drugs like XEN1101 could be the way forward.
9. Hot drug #2: STK-001
The second main strategy for genetic DEEs is to use advanced therapeutics like ASOs and gene therapies to tackle the genetic problem. That is what the ASO STK-001 from Stoke Therapeutics is doing: upregulating the mRNA for SCN1A so that patients with Dravet syndrome can have more of the ion channel. Dravet syndrome is largely caused by SCN1A haploinsufficiency so this is the ideal type of treatment to try to treat the entire syndrome.
Stoke initiated the current Phase 1/2 trial about three years ago, first with only a single administration to find tolerable levels, and then with multiple administrations of increasing doses. This is the first trial of its kind for DEEs, so it has allowed us to learn many things, like how long to wait before expecting improvement, but also which types of improvements and how large. The IEC2023 was the first large conference where Stoke presented their clinical trial data, after announcing the latest results this summer. And it is safe to say that they have finally found the good doses and the treatment is looking quite good. To quote Dr Dennis Lal recap of the data presentation:
“impressively, not only many patients have >50% seizure reduction but also cognitive measures show improvement.”
And that is what makes this treatment so special, the possibility to improve symptoms beyond epilepsy, in a way that can be quantifiable in clinical trials, because it is not acting on a symptom but on the disease itself.
Or as Dr Scott Perry from Texas said:
“finally a treatment showing evidence of true disease modification for Dravet syndrome!”
And I want to remind you that Dravet syndrome today, is many of the monogenetic DEEs in 5-10 years.
There is more to treatment development for DEEs than what we saw
At the last AES meeting I wrote that:
“We have crossed the line of developing treatments for symptoms, to developing treatments for the cause of the disease. And the line of focusing on a few syndromes only (those with the most patients), to seeing treatments in development for many more. That’s escape velocity.”
The IEC2023 meeting capture the first part very well, but missed the mark on the second. It captured very well the change in mentality and priorities of the clinicians and their representing organizations, perhaps even better than at AES, but barely scratched the surface of the true therapy development pipeline for DEEs, for many DEEs, so I recommend you to read my summary of the last AES meeting in this website, and see you in Orlando for AES2023.
I hope you enjoyed this update, let me know your thoughts in the comments,
Ana Mingorance, PhD
Disclaimer: I write these texts with the parents of people with rare epilepsy syndromes in mind, so excuse also my lack of technical accuracy in parts.

AES 2022: genetic epilepsies reach escape velocity
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. I review the progresses towards treating the cause of the rare epilepsies / DEEs.
PART 1 – ESCAPE VELOCITY
I often write a summary of the main lessons from the American Epilepsy Society meeting, but this year there was so so much about the rare epilepsy syndromes that I was not able to follow the main conference, there was barely enough time to follow all what was happening around the syndromes (which you also see referred to as Developmental and Epileptic Encephalopathies, or DEEs).
So I will focus my summary this year on the development of therapies for the rare epilepsy syndromes.
The conference started really strong, with the SYNGAP1 Conference the day before the main AES conference, where we learnt from Dr Jacquie French that for the first time, in 2022 we had more trials in rare epilepsies than in adult epilepsy. Even if people with rare diseases represent only about 20% of the total number of people with epilepsy, now they have the lion’s share of the trials. This is huge news!
And during the main AES meeting it was clear that indeed, much of the attention is currently on the rare epilepsies, with much of the agenda dedicated to genetic diagnostic, understanding how the adults with rare epilepsies look (so we can find them and develop treatments!), and much much research on the development of treatments. That’s why I believe that this year, with the milestone of having as many trials in syndromes as in regular epilepsy, we have crossed a line and likely reached escape velocity. Because it is not only about the two or three “famous” syndromes anymore, or about anti-seizure drugs being tested for the syndromes. We have crossed a different line.
At the other side of this line are treatments that target the cause of the monogenetic epilepsies. And those treatments look very different than classical anti-seizure drugs, and those trials also look different. We have crossed the line of developing treatments for symptoms, to developing treatments for the cause of the disease. And the line of focusing on a few syndromes only (those with the most patients), to seeing treatments in development for many more. That’s escape velocity.

PART 2 – TREATMENTS TO CORRECT THE CAUSE OF 8 SYNDROMES
Here are some of my highlights for eight of these monogenetic syndromes as an update to families. There are more diseases, don’t be discouraged if yours is not covered in this summary, I couldn’t do all of them.
SCN1A
Dravet syndrome is the (mostly) monogenetic syndrome that has received the most attention so far, and as a result the one with the most approved therapeutics. We saw many updates at AES about the approved drugs, and presentations about ongoing clinical trials with soticlestat (Phase 3) and EPX-100 (Phase 2). But I will focus on the treatments for the cause of the disease.
One of the most anticipated news of this congress were the early results from Stoke using an antisense oligonucleotide (ASO) called STK-001 to increase the good copy of the gene SCN1A. That’s because Dravet syndrome is a haploinsufficiency, which means that one copy of the gene is bad but one is good and can be exploited to produce enough protein from that one copy. That’s what Stoke is doing. After completing lower doses as part of the safety protocol for this type of therapy, Stoke has recently started administering higher doses to patients with Dravet syndrome and seen seizure frequency reduction and also improvements in non-seizure aspects of the disease. There is more information about this on their website. This is still the first small group of patients receiving enough dose to measure efficacy, and the results look very positive to me.
Another company using a similar antisense (ASO) approach, but directed to the SCN1A gene (not the RNA like Stoke), is CAMP4, that presented at AES how they are getting their antisense treatment ready for clinical trials (this is the antisense that OPKO was developing).
At the SYNGAP1 pre-meeting we saw a fantastic presentation from Tevard Bio about their approach to attack Dravet syndrome using different approaches all directed to the SCN1A RNA. And while Tevard still needs more time to get to clinical trials, Encoded Therapeutics is very close to starting trials with their gene therapy using a virus to increase SCN1A expression from the good gene copy, and presented at AES their work on finding good clinical scales to measure non-seizure symptoms of Dravet syndrome (the AES links are broken, so you will have to take my word for it). Encoded is preparing to start trials in very young children with Dravet syndrome (not yet started), and their gene therapy has the potential to change the developmental trajectory of these kids. The challenge is to get the right scales to capture that efficacy, so both them and Stoke have observational studies ongoing to validate those scales.
And Dravet syndrome also has at least two companies developing activators for the Nav1.1 channel, Lundbeck and Xenon. These are drugs, what scientists call small molecules, and not genetic therapies. From these two, Xenon has the most advanced program (another broken AES link that I cannot use), and I could see the Dravet syndrome space developing similar to SMA and have first an ASO approved, then a gene therapy, and finally a small molecule, all restoring sufficient levels of the sodium channel currents.
Shout-out to Veronica Hood, Scientific Director of the Dravet Syndrome Foundation who gave a fantastic talk about the impact of the patient groups in Dravet syndrome with major roles as conveners, educators, funders of early research and resource curators.
SCN2A
SCN2A is another sodium channel very important for neuronal functioning, so important that mutations that cause too much activity produce one of these developmental and epileptic encephalopathies (I will call it SCN2A Gain-of-Function) and mutations that cause one copy to not function also produce a syndrome with epilepsy and cognitive and behavioral manifestations (I will call it SCN2A Loss-of-Function). That means scientists need to find ways to turn off the channel or the gene for the first group, and find ways to boost expression of the good copy for the second group.
The company Praxis is doing just that, and working at both approaches with different types of treatment modalities. The most advanced program is called PRAX-562 and is a drug (a small molecule) designed to inhibit only two channels: Nav1.2 (that is the one produced by SCN2A) and Nav1.6 (produced by SCN8A). This drug looks really promising for people who carry mutations in either one of these channels that causes the channel to work too much, so it is for the Gain-of-Function mutations. And Praxis is about to start clinical trials for these two syndromes after completing Phase 1 in healthy volunteers.
What’s interesting is that Praxis also has a second program for people with SCN2A Gain-of-Function, and that is an ASO called PRAX-222 developed in collaboration with Ionis, and it is also getting ready to start clinical trials. So 2023 is going to be a very good year for SCN2A Gain-of-Function. And Praxis is also working on an ASO to do the opposite: increase SCN2A expression. This is a treatment for the SCN2A Loss-of-Function patents and it is still at earlier stages of development.
I will mention Praxis more times in this summary because they are working on more syndromes, and I want to highlight that this company has parents of kids with rare DEEs working with them as part of the team, and this is a very good sign. When we have scientists working within patient organizations, and patient advocates working within biotech/pharma companies, we can start breaking down many of the communication and cultural barriers between both worlds and developing better medicines.
SCN8A
SCN8A is the gene that encodes for another very important sodium channel, called Nav1.6. This is a channel that makes excitatory neurons fire, so the most common mutations are the Gain-of-Function type, that make those neurons way too active. What we need is clear: reduce the channel expression, or inhibit the channel activity.
This patient community has done a really good work at trial readiness, and we saw at least three therapeutic programs at AES (I might be missing some):
The clinical trial from Praxis with the drug that blocks Nav1.2 and Nav1.6 that is about to start.
A clinical trial with another drug, developed by Neurocrine and Xenon, that is only inhibiting Nav1.6. This one is already in clinical trials!
An ASO that we saw in posters and that reduces SCN8A expression so that there is not too much channel. This one is still in animal studies.
I look forward to the conference on SCN2A and SCN8A next spring because it looks like we will have many news about clinical trials for these syndromes!
KCNQ2
And from sodium channels we move now to potassium channels. This gene is very famous in epilepsy, because if you have a very young baby, under one month of age, that starts having seizures, the most likely cause is mutations in this gene. What these kids need is openers for this potassium channel, activators.
And there are two companies developing activators, one is Xenon, who are taking an older drug (ezogabine) that was taken off the market back into the market this time for KCNQ2 epilepsy. The drug is currently in Phase 3 studies, which means that if the trial is successful it will lead to an approval for KCNQ2. The second one is also an activator, from Knopp/Biohaven, that is currently in Phase 1 studies and will then move on to trials in patients with KCNQ2 epilepsy.
As with SCN8A, we have gone from not having trials to having even competing trials with drugs designed to correct the channel problem, either too much (Nav1.6) or too little (the potassium channel Kv7) activity. And I mainly look forward to seeing the potential benefit of these drugs past the very early years, once children with KCNQ2 mutations often stop having epilepsy but still have the developmental impacts that are due to the channel malfunction.
SYNGAP1
There is so so much going on for SYNGAP1 that I cannot summarize it all here. The pre-AES SYNGAP1 conference was fantastic, with many families in the room (and also all of the famous clinicians and many companies). There are iPSC models and mice and rats, I really liked the rat model and how it can be used to study drug response for seizures, sleep and cognition. They also have an idea of the SYNGAP1 isoform that is needed for gene therapies, and the “reversibility experiment” of putting SYNGAP1 back into adult mice has been done and it looks very positive. There is also a lot of work ongoing around patient data, like Ciitizen to see natural history, and an outcome measure for communication called ORCA being adapted to SYNGAP1 with funding from FDA. It’s all looking ready for therapies to be run through trials! There are two very strong SYNGAP1 foundations working to get trial ready and it shows.
And talking about therapies, there are several ASOs in development for SYNGAP1 and other modalities of gene therapies. What happens in this disease is the classical haploinsufficiency: one good copy of the gene, and one bad copy of the gene. That’s why the focus of these therapies is in promoting more expression from the good copy so that synapses can go back to working well again.
Praxis is working on an ASO to increase SYNGAP1. It is called PRAX-090 and in recent presentations Praxis says they hope to nominate a candidate in 2023. A “candidate” is when the company considers that their experimental drug or antisense is “good enough”, and at that point they stop tweaking it and start generating the animal safety data that is needed for trials, so it usually takes 1 to 1,5 years from the time of candidate nomination to starting trials.
Stoke Therapeutics, the company developing the ASO for Dravet syndrome (SCN1A) also has a program for SYNGAP1 using the very same approach, and they have partnered it with Acadia. And another company called CAMP4, that is also working on upregulating SCN1A for Dravet syndrome, presented at AES their early work towards also developing an ASO approach for SYNGAP1. And there are some additional academic and industry (Ionis) efforts to develop ASO treatments, so it is likely that SYNGAP1 families will get multiple options of clinical trials to increase protein levels, which should be a treatment for all the different symptoms of the disorder.
But antisense oligonucleotides are not the only approach to rescue SYNGAP1 expression. At the pre-AES conference we saw how Tevard is applying some of their gene therapies to increase expression from the SYNGAP1 RNA, and how scientists at Penn are using a CRISPR-like approach (called dCas9) to increase expression from the good gene copy. These are all very early stage efforts that are likely to reach clinical trials after the ASO treatments.
So you see, a lot going on, and not everything is presented at AES! And shout-out to Marta Dahiya, a clinician and SYNGAP1 mum who gave a very interesting talk during the main AES conference about digital natural history studies using platforms like the one from Invitae/Ciitizen.
STXBP1
Like the one before, STXBP1 is also a synaptic protein that when mutated causes haploinsufficiency: one good copy of the gene, and one bad copy of the gene. And the main therapeutic strategy to target the cause of the disease is therefore to promote more expression, often by exploiting the good copy, so that synapses can go back to working well again.
At AES there were several presentations around documenting the natural history of the disease, and also on validating scales to be able to run trials that count more than just seizures. The patient organization is working on a very large natural history study designed to validate outcome measures, and has recently announced natural history efforts by the companies Capsida and Encoded Therapeutics. These are gene therapy companies, the ones that use a virus to deliver the therapy to the brain, so we can imagine that gene therapies for STXBP1 are being developed even if they haven’t yet been announced.
There is also a known collaboration between the company Ionis and the Prosser lab to develop and ASO to restore STXBP1 expression, and at AES we also saw a presentation from the company Q-State about an antisense program to restore STXBP1 expression, which is still at early stages.
So it looks to me like STXBP1 disorder, like SYNGAP1 and others, have multiple companies working unannounced on therapies to correct expression of their faulty gene and that by next year AES there will likely be many more projects that have been officially announced. I very much look forward to that!
PCDH19
This is an interesting gene, because it encodes for a protein that makes cells touch each other to connect. It is in the X chromosome, so in females half of the neurons express the good X chromosome and the other half the one with the mutated PCDH19. That means that they have two type of neurons, with only half expressing the cell touch protein, and that is a problem because for neurons to touch each other they need to have the same code: either they both have PCDH19 or they don’t. What doesn’t work is only some having it. That is why in males, that express their only X chromosome in all neurons, mutations in PCDH19 don’t cause disease, because all neurons express the same code: no PCDH19.
There are at least two ASOs in development to make all neurons have the same cell touch code by eliminating expression of PCDH19 in all neurons in girls/women with PCDH19-Clustering Epilepsy. The first one is from Praxis, and is called PRAX-080 and as for SYNGAP1 the company has recently communicated that they hope to nominate a candidate in 2023 (the fully optimized ASO that then gets prepared to go to clinical trials). And the company Ionis is collaborating with the Parent lab to also develop an ASO that will achieve the same result: make female neurons like make neurons by knocking down PCDH19.
I like that for PCDH19 we have had clinical trials with small molecules (ganaxolone Phase 2) so we know for sure that seizures are countable and how a phase 2 trial could look. That helps de-risk the field for future trials, when companies will likely still count seizures as the main symptom for drug approval in addition to including scales to measure improvements in behavior and other domains – just as currently for Dravet syndrome.
CDKL5
CDKL5 Deficiency Disorder (CDD) is another DEE where the gene is in the X chromosome, but in this case the protein is a kinase and the problem is not that neurons don’t connect with different neurons, but that half of the neurons are missing the very important function of the protein CDKL5. The therapeutic goal is to put that protein back, with the added challenge that because only one CDKL5 copy is expressed in each neuron (only the good one or only the bad one) we cannot use many of the strategies used for haploinsufficiencies.
I dedicated a recent update to CDD from the CDKL5 Forum 2022, so please see that link to read how the first gene therapy trials to bring a new copy of CDKL5 to the brain of people with CDD might start as early as next year. And 2022 was a good year for CDD, we had the first drug approved for the disease: ganaxolone (approved by FDA, pending EU approval). So this year we had a new player in the big AES exhibit, with the large stand for Marinus highlighting their data for ganaxolone in CDD and helping educate the medical community about this rare epilepsy that now has a treatment.
As for PCDH19 epilepsy, CDD has very clear epilepsy that has been used already in clinical trials as the main measure for efficacy and that will help de-risk the upcoming clinical trials with therapies directed to correcting the cause, which should improve seizures as well as non-seizure symptoms (which are so much harder to measure). And like for other syndromes, what was talked at AES was just the top of the iceberg, and there are many more treatments in development for the cause of the disease that were not presented at the main medical conference. Check out the Forum update for that.

3 – IN CLOSING
If you are a rare disease parent you might have only read the section about your loved one’s disease gene. So I encourage you to do two things:
One, please remember that companies often don’t talk about their projects until they are close to trials, and not all present at AES, so what you read above is only a snippet of what’s going on in reality (iceberg!).
Two, whether you are a parent or a scientist, step back and look at all the syndromes and the global picture that emerges: I have only talked about the treatments in development to address the cause of the syndrome, not the symptom. Long gone are the times when there were no trials in the syndromes so we didn’t have good clinical evidence about anti-convulsant drug efficacy in them. And we are even closing chapter two, the one of cannabidiol and the other anti-convulsant drugs being tested and approved for syndromes, although their activity is not specific to the syndromes. What you are seeing here is different. You are seeing multitude of programs to block or open channels, to increase expression of good gene copies or reduce expression of bad ones, and several are already in clinical trials in patients. We are closing chapter two, and reading from chapter three… the one with treatments for the cause of the disease reaching clinical trials. And the speed at which this is happening, and how broadspread it is across different syndromes, is unstoppable. We have reached escape velocity.
Ana Mingorance, PhD
Disclaimer: I write these texts with the parents of people with rare epilepsy syndromes in mind, so excuse also my lack of technical accuracy in parts.

MAIN LESSONS FROM THE 2022 CDKL5 FORUM
For the past eight years the Loulou Foundation hosts an annual meeting where scientists and drug developers working on CDKL5 deficiency, together with representatives from patient organizations, meet to discuss the latest advances.
Here are the main news and take-home messages from the 2022 CDKL5 Forum that took place in November 7-8 2022.
For the past eight years, the Loulou Foundation has hosted an annual meeting, the CDKL5 Forum, where scientists and drug developers working on CDKL5 Deficiency Disorder (CDD), together with representatives from patient organizations, meet to discuss the latest developments in the field and to advance towards treatments and cures. You can find summaries from the past few meetings here: 2018, 2019, 2020 and 2021.

The 2022 CDKL5 Forum edition took place November 7-8 in Boston, returning to a face-to-face format after two on-line editions during the pandemic. Being back in person made this year’s meeting even more special. It was also, in my opinion, the best CDKL5 Forum meeting so far. Our understanding of CDKL5 deficiency biology and the pace of therapy development have advanced significantly in the last few years, so I will summarize my main take-home messages from this year’s Forum below and contrast them to where we were the last time we saw each other in Boston in 2019.
1. THE YEAR CDD BECAME PART OF THE 5 PERCENT
We always hear the same line: “there are more than 7,000 rare diseases, and only 5% have a treatment approved”. That is a special club of rare diseases that have received so much attention and investment that a medicine was specifically tested and approved for that rare disease.
In 2022, CDD joined that club.
In 2022, ganaxolone received approval from the FDA for treating seizures in CDD, and we are expecting the EMA decision soon. When we met last time at the 2019 Forum, we announced that Marinus was on track to complete recruitment for their ongoing Phase 3 trial with ganaxolone in CDD. Since we met last time, Marinus completed a first-ever global clinical trial for CDD in the middle of a global pandemic, and ganaxolone is now available for patients in the US and hopefully soon in other countries. I’m amazed at the courage of the Marinus team to bet on this un-tested rare disease, and to fight thought the pandemic to bring the first therapy to the market for people living with CDD. They have made the path easier for everyone else to follow.
And yet the first drug approval for CDD is only one of the two biggest news of the year…
2. CDD GENE THERAPY IS COMING TO CLINICAL TRIALS
One of the biggest hopes of the CDD patient families is the arrival of gene therapies to clinical trials. In CDD, half of the neurons (in girls) or all of the neurons (in boys) don’t produce the protein CDKL5, which is a very important protein for brain functioning. Gene therapies aim to use a virus to carry a new copy of the CDKL5 gene to neurons, so that they can start producing the protein [see also the next section in this update].
At the 2022 CDKL5 Forum, Emil Kakkis, CEO of Ultragenyx, said the words that CDD families were waiting for: “Gene therapy for CDD is coming to the clinic, we believe next year”.

Later in the conference, Sharyl Fyffe-Maricich (who has been leading the CDD gene therapy program at Ultragenyx) explained all of the steps they have taken to optimize their gene therapy to that it could get to as many neurons as possible before sitting down with regulators to talk about starting a clinical trial. The quality of their science is impressive, and Emil explained that clinical development is hard but we will all be in this together.
When we met in person last time at the 2019 CDKL5 Forum we were starting to have some data of experimental gene therapies in mice. Major questions back then were whether these therapies would work well enough in animals to progress to clinical trials, and when that might happen. We now finally have an answer and a timeframe: if everything goes well, clinical trials with the first gene therapy for CDD could start as early as in 2023.
This was the second big news of the year, and the highlight of the 2022 CDKL5 Forum.
3. TACKLING CDKL5 DEFICIENCY FROM ALL ANGLES
During the Forum Dinner, Phil Reilly (a very experienced developer of medicines for rare diseases) shared with us some stories about diseases that used to be considered untreatable until patient families intervened and played a key role to developing a cure. His message was: “there is no intractable disease”.
The next day, Emil Kakkis would remind us that “CDD has reversible components, and that is not true for many other neurological diseases”. He was referring to what we know from mice with CDD, where multiple aspects of the disease are reversible in older animals if CDKL5 is re-expressed. We also know that CDD is not a degenerative disease or even a strict neurodevelopmental disease where CDKL5 is needed only during a limited time in development. It is actually a neuromaintenance disorder, as we saw in the 2020 Forum, so there is much clinical hope around the possibility of restoring CDKL5 expression in CDD patients.

At the 2022 Forum we saw many updates about different approaches to tackle CDKL5 deficiency from all angles. Here are some quick notes on the different approaches:
Treat the disease symptoms: there is a Phase 3 clinical trial ongoing in CDD with the drug fenfluramine. This drug has beautiful efficacy in Dravet syndrome, and the company that developed it (Zogenix) was recently acquired by UCB Pharma, one of the leading epilepsy companies. The clinical trial is already enrolling in the US and expanding to Europe and Japan.
Treat the disease symptoms: Takeda had evaluated the efficacy and safety of soticlestat in a small study in CDD and updated us about the patients that continued taking the drug after the study. From the 12 patients that participated in the small trial, 8 are still taking the drug and all have experienced seizure reduction during the time they’ve been taking soticlestat. And it is not just about seizures, parents reported overall improvements including in communication and engagement. I hope to see this drug considered for treating patients with CDD.
Correct the CDKL5 pathway: there is a new discovery that opens the door to designing new treatments to correct some of the direct consequences of CDKL5 being missing. I dedicate to it the entire section 4 of this update.
Add more CDKL5 protein: the development of an enzyme replacement therapy for CDD (make the protein in the lab and give it to patients) has proven to be very challenging. Maria Luisa Tutino presented at the Forum her work towards producing full-length functional CDKL5 to be used for enzyme replacement and all of the challenges that they are solving one by one. There is need for more labs and companies to join the efforts to develop an enzyme replacement therapy for CDD because it remains an important therapeutic approach and it is very hard.
Correct the mutated RNA: the CDKL5 gene gets copied into messenger RNA, which then is used to produce the CDKL5 protein. If a gene has a mutation, then that mutation is still copied into the RNA. There are a variety of strategies to try to make functional protein from a mutated RNA without needing to fix the gene. For example there is a project ongoing to develop antisense oligonucleotides (ASOs) that could help with mutations in CDKL5 that cause splicing problems (like spinraza for SMA) or also that could potentially skip exons that might contain a mutation (skip part of the RNA sequence, like some treatments for Duchenne). And we know that there are also companies developing approaches to help cells read past non-sense mutations, which is sometimes also the problem for some people with CDD, so I hope to hear more news about these approaches and see them tested for CDD.
Reactivate the second CDKL5 gene copy: we had two presentations at the 2022 Forum about gene therapies that use a version of CRISPR to find the inactive CDKL5 copy and reactivate it. Kyle Fink has been leading the development of one of these gene therapies for CDD for the last couple of years, and recently was awarded a $1,4M grant to further develop the gene therapy for CDD. He also received the Lab of the Year award at the Forum.
Fix the mutated gene: there are several new approaches in development for correcting letters in a gene, or inserting letters. Majid Jafar, co-founder of the Loulou Foundation, explained that Loulou is missing one letter in her CDKL5 gene, and that her mum had asked “why can’t they just put that letter back?” when learning about the diagnosis. David Liu has been advancing a modified CRISPR-like approach called prime editing to do precisely that: put one letter back. He showed us how he can already fix the CDKL5 of kids like Loulou in cells in culture, and he is now working at getting it to work in living mice. Some of these gene editing approaches are starting to make it to clinical trials barely 5 years after the technology was discovered. His ultimate goal is not just to be able to add or replace one letter, but to replace the entire mutated CDKL5 gene by a healthy copy. It is always inspiring and a window into the future to hear David give a talk.
Add another copy of the CDKL5 gene: this is the classical gene therapy approach where scientists use a virus (usually AAV) to carry a gene to neurons. The virus cannot multiply, it just delivers the gene to neurons and from then on, neurons can produce the missing protein, in this case CDKL5. At the 2022 Forum we had a wonderful update about the Ultragenyx gene therapy program, and the news that it is progressing to clinical trials.
4. A BREAKTHROUGH IN UNDERSTANDING CDKL5
There were many presentations at the Forum about new understanding for how the CDKL5 protein works and some of the consequences to the brain when CDKL5 is missing. But I will only highlight one in this update, because I consider it a major breakthrough in our understanding of the protein and the disease. It also opens an important door to new treatments.
The discovery came from the lab of Sila Ultanir, at the Crick Institute. The protein CDKL5 is a “kinase”, which are proteins that can change the function of many other types of protein. This means that when the kinase is missing the problem multiplies because now you have all those other proteins not working well. The Ultanir lab has discovered a very important protein that needs CDKL5 to work properly, and it turns out to be a calcium channel (Cav2.3). When CDKL5 is missing, the calcium channel opens well but then closes too slowly, leading to too much calcium going through.
This is very important because many developmental and epileptic encephalopathies are caused by problems with ion channels, we call them “channelopathies” as a group. For example Dravet syndrome is caused by a sodium channel working less, KCNQ2 epilepsy is caused by a potassium channel working too much, and there is even a very rare syndrome caused by the Cav2.3 calcium channel working too much. The discovery that deficiency in CDKL5 makes that same calcium channel also work too much is unexpected and helps explain a lot.
I call this discovery a breakthrough for two main reasons:
1) We often wondered… if CDD is caused by a kinase missing, why does it look so similar to the channelopathies? Now we have the answer: CDD could be partly a channelopathy!
2) This discovery opens the door to designing new treatments to correct some of the direct consequences of CDKL5 being missing. Now we know that we want companies to develop Cav2.3 blockers.
5. MUCH PROGRESS EXPANDING THE RESEARCH TOOLBOX
For scientists to understand what CDKL5 does in cells, and to test treatments years before they are ready to go into trials, they need to model the disease in animals or cell cultures in the lab. There has been a giant effort by scientists across many countries to find animals or cells where we could remove CDKL5 and see a strong symptom (which scientists call “a phenotype”).
The most used animals in medical research are mice and rats. This year we had several great presentations that expand the CDKL5 research toolbox to fish and flies, and potentially frogs (tadpoles) and even pigs.

We had presentations from two groups showing that a tiny fish called zebrafish might help us research some of the symptoms of CDD. A group from Portugal has seen that zebrafish missing CDKL5 have bone problems (which also happens in patients!) and interesting motor symptoms that could be monitored to test experimental medicines in those fish. And a group from Boston is changing the fish genetics so that they can miss CDKL5 in only half of their neurons, which is what happens in girls with CDD.
My favorite presentations about new CDD models were two:
The lab of Ibo Galindo, in Spain, has made fruit flies that lack the CDKL5 gene and they have very strong epilepsy and their disease is so severe that they even die sooner than normal flies with CDKL5. In fact flies only have one gene for CDKL1/2/3/4 and 5, so these flies are missing all of the CDKL genes! And they look like other good fly models of genetic epilepsies. The flies also have interesting behaviors, and could be a powerful genetic system to understand the function of the 5 human CDKL proteins by making the flies express each of the human genes and see which ones can make them healthy.
The company Vyant is using stem cells taken from skin or blood samples from children with CDD to grow organoids in the lab. Organoids are like a ball of neurons and other brain cells all produced from the child stem cells and look like a mini-brain in a petri dish (they are to the right of the rat in the pictures that accompany this text). It turns out that organoids from kids with CDD have the in vitro equivalent of epilepsy. Vyant is using these organoids as a platform for drug screening directly in human cells, and they are already finding some promising compounds. I really liked their platform.
I was very impressed with the tenacity of so many scientists making a giant effort to leave no stone unturned, and to check every animal and every cell and tweak them and optimize them. This means that today we have a variety of different models that can be used to answer different questions about biology and treatments, and this type of work takes a big community effort, it cannot be done by only a few labs. I’ll come back to this in the next section.
6. “WE ARE IN THIS TOGETHER”
A theme that emerged as the 2022 CDKL5 Forum motto was “we are in this together”. We heard it in words, and we saw it in action. These are only some of the examples that we saw during the Forum:

No patient left behind. There are patients with CDD in all countries, and that also means they come in all languages. Sadly language barriers are a major limitation to reach the international community. To bring together the data of as many patients as possible, and to be able to reach out to them about news such as clinical trials, the CDKL5 Registry is recently available in many of the common languages and will be expanding to more. Current languages are English, French, German, Italian, Spanish, Arabic, Chinese (traditional and simplified), Japanese and Russian! This was a lot of work, but we are in this together.
Companies collaborating in clinical studies. When we met last time in Boston in 2019, we talked about the need for an observational study to validate outcome measures for CDD (to check that we know how to track CDD symptoms in a way that is useful for clinical trials). The catch is that we wanted only one study, with the different pharma and biotech companies coming together to co-design it and run it, so that we would not overburden the patient community. At the 2022 Forum, Xavier Liogier from the Loulou Foundation stood in that same stage to announce that the first patient has been enrolled in that study, called the CANDID study, which will have the size of a Phase 3 trial in CDD. The years in between came with much work (including getting the study reviewed by the FDA!) and also with a giant step from seven companies that came together in a study that is a first-of-a-kind precompetitive collaboration. We are in this together.
Preclinical collaborations. The CDKL5 research community has always been exceptionally collaborative, and the Forum has a series of parallel workshops (the last two editions also added pre-meeting workshops) where we get organized. At the 2022 Forum we saw a proposal to get the companies developing treatments for CDD more actively involved in this preclinical collaboration network, because it is easy for pharma and biotech scientists to share advice, but hard to share actual research data! The goal is to have academics and companies compare their CDD mouse data to validate what are the best and most reliable endpoints (the best mouse symptoms). The academic groups are already doing this, and I hope that by next year’s Forum we can announce that companies are also joining this very needed effort. We are in this together.
The patient alliance helps accelerate clinical studies. A big challenge for companies to run trials in rare diseases is to know how to find the right hospitals. The CDKL5 Alliance has put together a list of hospitals in the different Alliance countries where they have trusted clinicians. We learnt at the Forum that this resource has already helped accelerate the initiation of the observational CANDID study, and I hope that it will also help accelerate treatment trials. We are in this together.
The leadership of patient families. The CDKL5 Forum is a scientific and medical conference, where we review the news from the previous year and get organized to help progress the field further. But it is also a meeting led by a patient family, where patient families feature prominently in the agenda and the discussions. We opened the meeting with the words from Natalie Ladly from CDKL5 Canada, sharing with the audience the toll of CDD on her family (#brynnstrong!). Simon and Fiona Walsh from CDKL5 Ireland also invited us to look into their family’s life during the Forum Dinner, and Rick Upp, from the IFCR, closed the meeting with an update from the CDKL5 patient community. The Champion of Progress CDKL5 Forum Award of this year went to Antonino Caridi, from CDKL5 Italy, one of the most loved and respected leaders from the patient community in the fight for a cure for CDD. And representatives from 14 different countries (the Alliance includes more than 30 countries!) were also in attendance, mixing with researchers and actively participating in the discussions, in particular during the interactive breakout sessions. That’s how the words and actions from the patient families during the Forum also spelled out how we are in this together.

SUMMARY
If we had to boil down the 2022 Forum to a few sentences, it would look like this:
There is no intractable disease.
CDD is now part of the 5% of rare diseases that has a treatment approved.
CDD gene therapy is coming to clinical trials.
And clinical development is hard, but we are in this together.
The Loulou Foundation started in 2015 with the mission to have “treatments for CDD (in clinical trials) by 2020, and cures by 2025”. Back then there were not therapeutic programs at all being developed for CDD, but there was faith in science and in the power of a large community. With the news shared at the 2022 Forum, it now seems realistic to believe that even before 2025 there will be people living with CDD who will have received a gene therapy as part of a clinical trial. This was a great year for CDKL5 research.
I hope you enjoyed this summary. Please let me know your thoughts in the comments.
Ana Mingorance, PhD
Disclaimer: These are my own impressions from the presentations and topics that I was most interested in as a scientist and supporter of the patient community, and not an official text about the Forum by the Loulou Foundation. I write these texts with the parents of people with CDD in mind, so excuse also my lack of technical accuracy in parts.

REPASO DEL FORO CDKL5 2022
La octava edición del Foro CDKL5 tuvo lugar en Boston, los días 7 y 8 de noviembre. El Foro es una reunión anual que organiza la Fundación Loulou y en la que científicos y miembros de la industria farmacéutica se reúnen con representantes de la comunidad de pacientes para repasar los últimos avances en el campo.
Este es un repaso para los grupos de pacientes de las principales novedades del Foro CDKL5 2022.
Hace ya ocho años que la Fundación Loulou organiza una reunión anual, el Foro CDKL5, donde los científicos de academia y de industria trabajando en el síndrome de deficiencia en CDKL5 (CDD), junto con representantes de los grupos de pacientes, se reúnen para compartir las últimas novedades y avanzar hacia tratamientos y una cura.

La edición de 2022 tuvo lugar los días 7 y 8 de noviembre en Boston, volviendo al formato presencial tras dos años en formato virtual debido a la pandemia. Eso ha hecho el Foro de 2022 aún más especial. Para mi este ha sido el mejor Foro desde 2018 que fui al primero. Ha habido un progreso enorme en lo que sabemos de CDKL5 y la velocidad de progreso de las terapias, así que voy a resumir las principales noticias del Foro de 2022 y compararlas con cómo estaban las cosas la última vez que nos reunimos en persona, en 2019 también en Boston.
1. EL AÑO EN QUE NOS UNIMOS AL CLUB DEL 5 PORCIENTO
Seguro que habéis oído eso de “hay más de siete mil enfermedades raras, pero solo el 5% tiene algún tratamiento aprobado”. Es un club especial ese 5%, son las enfermedades raras que han recibido suficiente atención e inversión para que se haya desarrollado y llevado a ensayos clínicos un tratamiento específicamente para ellos.
En 2022, CDD se unió a ese club.
En 2022 ganaxolona se aprobó en Estados Unidos para tratar las crisis epilépticas en CDD, y estamos a esperas de las decisión de la agencia europea que se espera en cuestión de meses. Cuando nos vimos por última vez en Boston en 2019 hablábamos de que Marinus estaba cerca de completar el reclutamiento para su ensayo clínico de fase 3 con ganaxolona en CDD. Desde entonces Marinus completó el reclutamiento en un ensayo por primera vez global en CDD, en medio de una pandemia también global, y ya el fármaco está aprobado y disponible para los pacientes en Estados Unidos y esperamos que pronto en otros países. Hay que destacar la valentía del equipo de Marinus de apostar por una enfermedad que no tenía tratamientos previos, y de luchar a través de una pandemia para sacar a mercado la que sería la primera terapia para CDD. Han allanado el camino que ahora es más fácil para los que vienen detrás.
Y resulta que la aprobación del primer fármaco para CDD solo es una de las dos grandes noticias del año…
2. LA TERAPIA GÉNICA PARA CDD AVANZA A ENSAYOS CLÍNICOS
Una de las mayores esperanzas para las familias CDKL5 es la llegada de las terapias génicas a ensayos clínicos. En CDD, a la mitad de las neuronas (en niñas) o a todas (en niños) les falta CDKL5 por tener un gen mutado. Y eso les impide producir la proteína CDKL5 que es muy importante para el funcionamiento neuronal. Las terapias génicas buscan usar un virus para llevar una nueva copia del gen CDKL5 a las neuronas, y que así puedan volver a producir la proteína [ver el repaso de estrategias de tratamiento en la sección siguiente].
En el Foro 2022, Emil Kakkis, que es el director ejecutivo de Ultragenyx, por fin dijo las palábras por las que llevábamos esperando los últimos años: “la terapia génica para CDD viene a ensayos clínicos, creemos que el año que viene”.

Más tarde vimos también la ponencia de Sharyl Fyffe-Maricich que es quien lidera el proyecto de la terapia génica de CDKL5 en Ultragenyx, y nos explicó todos los pasos que han tomado para asegurarse de que su terapia génica llega a tantas neuronas como sea posible antes de sentarse con la agencia reguladora a hablar de empezar ensayos clínicos. La calidad de la investigación que hace esta gente es impresionante, y Emil nos explicó que el desarrollo de una terapia a través de ensayos clínicos siempre es difícil pero que estamos juntos en todo esto.
La última vez que nos vimos en Boston en 2019 vimos la presentación de datos de terapias génicas en ratones con deficiencia en CDKL5, y en ese momento nos hacíamos aún dos preguntas muy importantes: si llegarían a funcionar suficientemente bien como para avanzar a ensayos clínicos, y cuando sería. Ahora por fin ya tenemos respuestas y fechas orientativas: si todo va bien, los primeros ensayos con la primera terapia génica para deficiencia CDKL5 podrían empezar en 2023.
Esta es la segunda de las grandes noticias del año, y sin duda el notición del Foro.
3. ATACANDO A CDKL5 POR TODOS LOS LADOS
Durante la cena de recepción del Foro, Phil Railly (un descubridor de terapias para enfermedades raras muy importante) nos compartía varias historias de enfermedades que se consideraban intratables hasta que padres de niños afectados tomaron cartas en el asunto y jugaron un papel clave para desarrollar curas. Su mensaje era que “no hay enfermedades intratables”.
Luego al día siguiente Emil Kakkis nos recordaba que “CDD tiene aspectos reversibles, y eso no pasa en muchas otras enfermedades neurológicas”. Hacía referencia a lo que hoy en día sabemos de que en ratones con deficiencia en CDKL5 la enfermedad es reversible, y también sabemos que no es una enfermedad degenerativa, ni siquiera de las típicas de neurodesarrollo donde la falta de la proteína durante etapas claves de desarrollo causa daños irreversibles. Hoy entendemos que la deficiencia en CDKL5 es un desorden de “mantenimiento de función neuronal”, como vimos en el Foro de 2020, con lo que hay mucha esperanza de cara a la posibilidad de devolver la expresión de CDKL5 a los pacientes con CDD.
En el Foro 2022 vimos actualizaciones sobre muchas aproximaciones terapéuticas para atacar a la deficiencia en CDKL5 por todos los lados. Os hago un resumen de lo que vimos:

Tratar los síntomas: el ensayo con fenfluramina está en marcha y es el segundo ensayo de Fase 3 que tenemos en CDD. Fenfluramina ha tenido resultados muy buenos en el síndrome de Dravet, y la empresa que la desarrolla (Zogenix) ha sido comprada por una de las mayores empresas farmacéuticas líder en el campo de epilepsia, UCB Pharma. Los ensayos clínicos están reclutando en Estados Unidos y pendientes de arrancar en Europa y Japón.
Tratar los síntomas: Takeda evaluó la eficacia y tolerabilidad de soticlestato en un estudio pequeño en CDD, y nos trajo una actualización de 8 de esos 12 niños que se quedaron tomando el fármaco tras terminar el ensayo. Todos han experimentado mejoría en el control de crisis y los padres reportan mejoras importantes en otros dominios como atención y comunicación, con lo que esperamos que Takeda considere CDD como otra indicación para soticlestato.
Corregir los problemas en las otras proteínas: si sabemos qué proteína no funciona bien al faltar CDKL5 podríamos ir directamente a arreglarlas y compensar por la falta de CDKL5. En este punto hubo un descubrimiento muy importante este año, al que dedico la sección siguiente entera.
Aportar más proteína: el desarrollo de terapias de reemplazo enzimático para CDD (cuando produces la proteína en el laboratorio para dársela a los pacientes) ha resultado ser tremendamente difícil. Maria Luisa Tutino presentó en el Foro su trabajo para conseguir la producción de proteína CDKL5 funcional en el laboratorio para usarla como terapia, y los desafíos con los que se iba encontrando y que van solucionando uno tras otro. Yo espero que se unan más investigadores y empresas a intentar conseguir una terapia de reemplazo enzimático para CDD porque hace mucha falta y es muy complejo.
Arreglar el ARN mensajero: el gen CDKL5 se copia en ARN mensajero, y este a su vez sirve de instrucción para hacer la proteína CDKL5. Hay varias estrategias para intentar sacar proteína funcional a partir de un ARN mutado, y en el Foro vimos una estrategia en Boston para hacer oligonucleótidos antisentido (ASOs) que es un tipo de terapia disponible en otras enfermedades genéticas. Estos ASOs servirían para ciertas mutaciones en CDKL5, no todas, por ejemplo para intentar leer el ARN cuando hay mutaciones de “splicing” o si hay algún cacho de CDKL5 menos importante, haciendo que la célula se salte la lectura de ese trozo del ARN (salto de exón). Hay ejemplos de estas estrategias en la atrofia muscular espinal y en Duchenne, pero para CDKL5 apenas están empezando.
Reactivar la segunda copia del gen CDKL5: tuvimos dos presentaciones de grupos que buscan usar una especie de CRISPR para llegar a la copia inactiva de CDKL5 (en niñas) y hacer que se exprese. El invesitgador Kyle Fink lleva años liderando uno de esos proyectos y le acaban de conceder cerca de un millón y medio de dólares para avanzar su terapia génica para CDD (que hace que se lea la segunda copia). Esta año recibió el premio al mejor Laboratorio del Año en el Foro CDKL5.
Arreglar el gen mutado: hay varias estrategias en desarrollo para corregir las letras en un gen, o para poner una letra que falta. Majid Jafar, co-fundador de la Fundación Loulou, nos explicaba que a Loulou le falta una letra en el gen CDKL5 y que al saber el diagnóstico su madre se preguntaba “¿y por qué no le ponen la letra que falta y ya está?”. Pues el investigador David Liu está usando también una especie de CRISPR que se llama “prime editing” para hacer justamente eso, poner las letras que faltan. Nos enseñó cómo ya pueden arreglar el gen CDKL5 de niños como Loulou en células en el laboratorio, y actualmente están trabajando en hacer que funcione en ratones. Este tipo de edición genética está empezando ensayos clínicos en otras enfermedades y se descubrió hace apenas 5 años. El objetivo de David no es ya solo poner o quitar una letra, sino como nos dijo “cambiar el gen entero por una copia buena”. Las presentaciones que da este hombre son siempre una ventana hacia el futuro.
Aportar una copia nueva del gen: esta es la terapia génica clásica, en la que los científicos usan un virus (generalmente uno llamado AAV) para llevar el gen que falta a las neuronas. Ese virus no se multiplica, sino que sirve para llevar el gen a las neuronas y que a partir de ese día ya puedan producir la proteína, en este caso la de CDKL5. En el Foro 2022 tuvimos el notición de Ultragenyx de que su terapia ahora avanza hacia la fase de ensayos clínicos.
4. UN DESCUBRIMIENTO MUY IMPORTANTE PARA ENTENDER CDKL5
Vimos muchas presentaciones en el Foro sobre entender mejor cómo funciona la proteína CDKL5 y las consecuencias de su falta para el cerebro. Pero solo voy a hablar de una en este resumen, porque es un descubrimiento fantástico que ayuda a entender lo que hace la proteína y lo que pasa en la enfermedad. También es una puerta abierta a diseñar nuevos tratamientos.
El descubrimiento nos llega del laboratorio en Londres de Sila Ultanir. La proteína CDKL5 es una kinasa, que son proteínas que lo que hacen es modular la función de muchos otros tipos de proteína. Esto multiplica los problemas, porque cuando falta la enzima tienes también todas esas proteínas que no funcionan bien. El grupo de Sila ha descubierto una proteína muy importante de esas que necesitan a CDKL5 para funcionar bien, y resulta ser un canal de calcio (Cav2.3). Cuando falta CDKL5, ese canal de calcio de abre bien pero luego se cierra muy despacio, lo que lleva a demasiado calcio pasando por el canal.
Estos es muy importante porque muchos de los síndromes de neurodesarrollo con epilepsia están causados por problemas con canales iónicos, las llamamos las canalopatías. Por ejemplo el síndrome de Dravet es por falta de canales de sodio, y la epilepsia por KCNQ2 es por mutaciones que dan demasiada actividad a canales de potasio. Curiosamente hasta hay un síndrome ultra-raro causado por tener el canal de calcio Cav2.3 demasiado abierto, y eso es justamente lo que han visto que pasa también cuando falta CDKL5. Esto era inesperado, pero explica muchas cosas.
Y digo que es un descubrimiento muy importante por dos razones:
1) Muchas veces nos preguntamos… si CDD está causado por faltar una kinasa por qué se parece tanto a algunas canalopatías. Ahora sabemos la respuesta: ¡CDD es parcialmente una canalopatía!
2) Este descubrimiento abre las puertas a diseñar nuevos tratamientos dirigidos a corregir las consecuencias de la falta de CDKL5. Ahora sabemos que queremos que alguna empresa desarrolle un fármaco bloqueante de Cav2.3.
5. MUCHO PROGRESO AMPLIANDO LA CAJA DE HERRAMIENTAS DE INVESTIGACIÓN
Para poder entender qué hace CDKL5 y probar terapias antes de que lleguen a ensayos, los científicos necesitan modelos de la enfermedad en animales o en células en cultivo. Y ha habido un esfuerzo de titanes por parte de investigadores en muchos países para encontrar animales o tipos de cultivo celular al que la ausencia de CDKL5 le provoque síntomas claros (lo que llamamos fenotipos).
Y en investigación casi siempre se usan ratones y ratas modificados genéticamente para tener el mismo problema que la enfermedad en humanos. Pero este año vimos presentaciones de equipos que están ampliando la caja de herramientas de investigación de solo ratones y ratas a incluir muchos otros, como peces, moscas, renacuajos y hasta cerdos con deficiencia en CDKL5.

Vimos ponencias de dos equipos usando peces cebra como modelo de deficiencia en CDKL5, uno de Portugal y uno de Boston. Los peces tienen algunos de los problemas que vemos en personas afectadas, por ejemplo problemas óseos, y están buscando hacer peces a los que les falte CDKL5 solo en la mitad de las neuronas que es lo que pasa en niñas y por tanto es la mayoría de los casos.
Mis presentaciones favoritas sobre modelos de deficiencia en CDKL5 fueron dos:
El laboratorio de Ibo Galindo, en Valencia, ha hecho moscas de la fruta (o mosca del vinagre) con deficiencia en CDKL5 y visto que desarrollan epilepsia y su enfermedad es tan severa que hasta viven menos que las moscas sin modificar. Lo curioso es que las moscas solo tienen un gen CDKL, en vez de cinco como las personas, con lo que a estas moscas ¡les falta el equivalente de CDKL1/2/3/4 y 5! Y les produce los mismos síntomas que tienen moscas de otras epilepsias genéticas, las de CDKL5 incluso tienen cambios en su comportamiento. Las moscas de la fruta son de mucha utilidad como herramienta de investigación genética, porque se le pueden quitar y poner genes y ver su función, o en este caso darles los genes humanos de los cinco CDKL y ver cuales les corrigen la enfermedad.
La empresa Vyant está usando células madre de niños con CDKL5 para producir organoides. Los organoides son como una pelota de neuronas y otras células cerebrales todas producidas a partir de las células madre que se sacan de la piel o muestra de sangre con lo que llevan el ADN de los niños, y que forman en cultivo como un mini-cerebro (en la foto son la bola que está a la derecha del todo). Pues resulta que los organoides de niños con CDKL5 tienen la versión in vitro de epilepsia, que son descargas eléctricas hipersincronizadas. Y lo están usando como plataforma de cribado de fármacos y ya han encontrado algunos compuestos.
Lo que más me impresiona es la tenacidad de tantos científicos haciendo un esfuerzo de gigantes para no dejar piedra sin remover, mirando en cada animal y cada tipo de cultivo celular y buscando la forma de encontrarle la señal de la falta de CDKL5. Gracias a eso hoy tenemos un abanico de modelos animales y celulares que permiten hacer muchas preguntas científicas, tanto de biología como de tratamientos, y este tipo de trabajo requiere un gran esfuerzo de grupo, no lo puede hacer un laboratorio solo. Y de eso trata la sección siguiente de este resumen.
6. “ESTAMOS EN ESTO JUNTOS”
Hay un mensaje que se convirtió en el lema del Foro 2022, y es “estamos en esto todos juntos”. Lo oímos en palabras pero también lo vimos en acciones. Estos son solo algunos de los ejemplos que vimos durante el Foro:

Que no se quede nadie atrás. Hay personas con CDD en todos los países, y eso implica también que son personas con todos los idiomas. Por desgracia la barrera lingüística es una de las principales barreras a la hora de llegar a la comunidad internacional de pacientes. Para poder reunir datos de tantos afectados por CDD como sea posible, y poder mantenerles informados de noticias como ensayos clínicos, el registro CDKL5 Registry está disponible desde hace poco en la mayoría de los grandes idiomas: Inglés, Español, Francés, Alemán, Italiano, Árabe, dos versiones de Chino, Japonés y Ruso. Ha sido un trabajazo pero estamos en esto todos juntos.
Empresas colaborando en estudios clínicos. Cuando nos vimos la última vez en Boston en 2019 lanzamos la propuesta de que hacía falta hacer un estudio clínico en CDD para validar escalas clínicas (para verificar que sabemos medir los síntomas de CDD de la forma que hace falta para ensayos clínicos). Y lo que pedíamos no era simple, porque nuestra propuesta es que fuera solo un estudio en el que colaboraran todas las empresas, co-diseñándolo y ejecutándolo juntas, para así no duplicar (o quintuplicar) esfuerzos y sobrecargar a las familias. Este año Xavier Liogier, de la Fundación Loulou, nos hablaba desde ese mismo escenario pero esta vez para anunciar que ya ha entrado el primer paciente a ese estudio clínico, llamado CANDID, que incluye muchos países incluida España. Entre esas dos ponencias ha habido muchísimo trabajo, incluso llevamos el estudio a examen por la agencia americana del medicamento, y también ha supuesto un importante paso hacia la colaboración por parte de las 7 empresas que se juntaron para hacer el estudio posible. Estamos en esto todos juntos.
Colaboraciones preclínicas. La comunidad de investigadores en CDKL5 siempre ha sido excepcionalmente colaborativa, y el Foro tiene una serie de sesiones de grupo en la que nos coordinamos. Este año salió la propuesta de que las empresas que están buscando terapias para CDKL5 se unan a esas colaboraciones preclínicas (de investigación en animales) porque suelen estar presentes en la conversación pero lo que no hacen es abrir sus libros de laboratorio y enseñarnos los datos. Lo que se busca es que los investigadores trabajando con ratones con CDD en universidades y en empresas se puedan sentar juntos y comparar resultados y poder así validar los mejores síntomas en ratones que podemos usar para investigar (los síntomas más robustos y mas reproducibles entre laboratorios). Los grupos académicos ya lo hacen y el objetivo es que se unan los de las empresas. Espero que el año que viene en el Foro 2023 podamos confirmar que hemos abierto la colaboración y que se han unido a un esfuerzo tan necesario, porque estamos en esto todos juntos.
La Alianza internacional ayuda a acelerar estudios clínicos. Una dificultad habitual de los ensayos clínicos en enfermedades raras es que les cuesta mucho a las empresas identificar los hospitales que hacen falta. La Alianza Internacional de CDKL5 ha compilado una lista de los hospitales de cada país de la alianza en los que los miembros tienen médicos de confianza con experiencia en CDD. En el Foro aprendimos que esta lista ha facilitado y acelerado la identificación de hospitales para el estudio CANDID, y espero que también acelere los futuros ensayos clínicos. Estamos en esto todos juntos.
El liderazgo de las familias CDKL5. El Foro CDKL5 es una conferencia médica y científica, donde nos reunimos para repasar los últimos avances y para organizarnos de cara a seguir avanzando. Pero también se trata de un congreso liderado por los padres de una niña con CDD, donde las familias tienen un papel prominente tanto en la agenda como en las conversaciones. Abrimos el Foro con las palabras de Natalie Ladly, de CDKL5 Canada, que compartió con todos nosotros lo que supone para su familia vivir con CDD (#brynnstrong!). Luego durante la cena de gala Simon y Fiona Walsh de CDKL5 Irlanda también nos abrieron las puertas de su casa y su familia, y para cerrar el congreso Rick Upp, de la IFCR, nos hizo un resumen de lo que ha pasado este último año en la comunidad de pacientes. El premio al “Paladín del Progreso” que da el Foro cada año a las personas que más impacto han tenido en CDKL5, fue este año a Antonino Caridi, de CDKL5 Italia, uno de los líderes mas querido y respetado de la comunidad de pacientes en su lucha por una cura para CDD. Y tuvimos representantes de pacientes de 14 países (¡ya hay más de 30 en la Alianza!) que participaron en el Foro, mezclándose con investigadores y participando de manera activa en las conversaciones, sobretodo durante las sesiones de debate. Es así que a través de sus palabras y sus acciones las familias CDKL5 nos enseñaron durante el foro que estamos en esto todos juntos.

RESUMEN
Si se puede resumir el Foro de 2022 en unas pocas frases, sería:
No hay enfermedades intratables.
CDD es ahora parte del 5% de enfermedades raras con un tratamiento aprobado.
La terapia génica viene a ensayos clínicos.
Y desarrollo de una terapia a través de ensayos clínicos siempre es difícil pero estamos en esto todos juntos.
La Fundación Loulou fue fundada en 2015 con la misión de tener “tratamientos para CDD en ensayos en 2020, y curas en ensayos en 2025”. En aquellas fechas no había ni un solo fármaco en desarrollo para CDD, pero si que había fe en la ciencia y en el poder de crear una comunidad en torno a una enfermedad rara. Tras las noticias del Foro 2022 ahora ya sabemos que es realista pensar que incluso antes de 2025 habrá personas que hoy viven con CDD que habrán recibido una terapia génica como parte de esos ensayos clínicos. Este ha sido sin duda muy buen año para la investigación en CDKL5.
Espero que os haya gustado el resumen. Ya me diréis lo que os parece en los comentarios.
Ana Mingorance, PhD
Nota: este texto captura mis impresiones de las presentaciones del Foro que más me interesaron como científico y como defensora de los pacientes, no es un texto oficial del congreso emitido por la Fundación Loulou. Escribo estos resúmenes para los padres de personas con CDD, así que a veces me tomo ciertas licencias a la hora de explicar las partes mas técnicas.

Top 5 insights from the American Epilepsy Society meeting (2021)
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. These are my top 5 insights from the American Epilepsy Society 2021 meeting.
The American Epilepsy Society (AES) meeting is the largest epilepsy meeting of the year, and because it takes place every month of December it also serves as an annual review on the understanding and treatment of epilepsies. I look forward every year to the first week of December for this reason.
I look for therapies for rare genetic epilepsies, and in the recent years this area has exploded to take over much of the AES meeting. Because of that, I will focus this update on the rare genetic epilepsies, or Developmental and Epileptic Encephalopathies (DEEs). There is a lot more that is presented at the AES meeting and that I will not cover. These are my top 5 insights from the American Epilepsy Society 2021 meeting.
1. Natural History Studies are tremendously important
When companies develop treatments for seizures as a symptom, it is straightforward to run trials: just count seizures. But when companies start developing treatments that treat the cause of that epilepsy, and in particular when we are talking about complex neurodevelopmental syndromes with epilepsy, then clinical trials are more complex than just counting seizures, and need more solid data on the usual symptoms and severity that those patients have so that we can know if there is efficacy. This is where Natural History Studies (a type of observational study) specifically designed to inform on clinical trial design come into play. And we are seeing a big number of these.
Several of these ongoing studies were presented at the AES meeting. The BUTTERFLY study by Stoke Therapeutics is an observational study to characterize clinical scales to measure the non-seizure aspects of Dravet syndrome, and has already shown promise with several scales that could be used in interventional clinical trials. The ENVISION study by Encoded Therapeutics, has also helped validate scales and determine the baseline characteristics on non-seizure outcomes in young children with Dravet syndrome. Both companies are developing treatments to rescue SCN1A gene expression in Dravet syndrome, so these observational studies are designed to make those treatment trials possible, and both observational studies are already producing very useful data.
We also saw example of more proactive observational studies that are already ongoing even before companies are ready for trials, like an SCN8A Natural History Study resulting from a partnership between hospitals and patient groups, and a consortium working on STXBP1 Natural History. While these studies are simpler than the company-run Dravet syndrome studies, and don’t validate scales, they help us understand the collection of symptoms and their severity in these syndromes, and guide the design of the clinical-trial enabling studies. It is also remarkable that both studies include data from more than 500 patients from each syndrome!
If you are running or supporting a rare epilepsy patient group, I recommend you also check these two additional resources to understand why these studies are so important and consider how it could work for your disorder:
1) The pre-competitive industry collaboration involving seven biopharmaceutical companies and the Loulou Foundation to run an observational study specifically designed to evaluate the feasibility and suitability of a collection of clinical outcome measures in CDKL5 deficiency: here and here.
2) The amazing Mike Granglia from SYNGAP Research Fund explains in a recent video why Natural History Studies specifically designed to inform on clinical trial design are so so important to not delay the initiation of clinical trials for these syndromes. Please watch it, the Natural History Studies part starts around minute 5:45 but the entire videos are fantastic.
2 – Gene therapies are coming for many epilepsies
There are many reasons to believe that many of the genetic epilepsies will benefit much from restoring gene expression in patients, even in older individuals. And we saw several studies in mice presented at AES where scientists returned gene expression to mice with genetic epilepsies showing very good efficacy. See for example this study in CDKL5 Deficiency Disorder, this one in SCN1A Dravet syndrome, and this one in STXBP1-related DEE.
To be able to do that in patients, scientists need to develop gene therapy approaches using antisense oligonucleotides (ASOs) or virus that can bring to the brain the missing gene, or virus that can bring to the gene CRISPR approaches or transcription factors (see review of options for Dravet syndrome here). We saw several of this presented at AES this year.
The most advanced program is the one from Stoke Therapeutics. In addition to presenting an update about their observational study, Stoke presented new information about how their ASO to increase SCN1A distributes within the brain in non-human primates, and the likely doses that are needed to produce enough sodium channel in the brain in trials. From their data, it seems that repeated administration of 30mg of STK-001 (their ASO to increase SCN1A levels) will be a good target to to achieve active brain levels in patients. They are not yet administering those levels to patients because the interventional trial is still at early stages, and it is a Phase 1/2 trial without any prior trial in healthy volunteers, therefore starting with only one administration of STK-001 to Dravet patients at very low dose and moving from there to higher doses, still with only one administration to monitor safety, and then starting with repeated dosing also from lower concentrations to higher. From this early data, Stoke reports good tolerability, and a trend towards having less seizures but all participants had received either only one administration of STK-001 or multiple administrations of 20mg, so we really have to wait for the trial to reach higher dose with repeated administration in order to see the therapeutic potential of this treatment. We might potentially see this first from their UK study, called ADMIRAL, where Stoke will evaluate multiple doses of up to 70mg [see Note for parents at the end of this text].
Behind STK-001, there are several therapies in development, all designed to increase SCN1A for Dravet syndrome, that have not yet started clinical trials. You can see a summary of all those approaches here, where I explain the biology of SCN1A deficiency and the options to increase it.
Encoded Therapeutics is developing ETX101, a gene therapy that uses an AAV virus to upregulate SCN1A in the brain. They are preparing to start clinical trials, and this year presented data on their observational study as well as on understanding caregiver perspective to better design their upcoming clinical trial. A company called CAMP4 has acquired the rights to the ASO program from OPKO to upregulate SCN1A, and talked about the preclinical data that is available for this program at AES. And there are at least three academic groups developing viral-based gene therapies for Dravet syndrome, all focused on increasing the SCN1A gene: the adenovirus program that we saw last year at AES, a new one this year, and a CRISPR-ON approach being developed in Italy.
We also saw preclinical data for a gene therapy program for SLC13a5 Deficiency, showing efficacy in young mice and also in young adults, which is very promising towards future studies. And scientists at UCL in the UK are working on bringing gene therapy to non-genetic epilepsy! Their gene therapy is designed to sense if neurons are hyperactive, turn on and produce potassium channels (which makes neurons less excitable), and as soon as neurons are not hyperactive anymore, turn off. This means that neurons would only make use of that gene therapy when they truly need it. They are still trying it in mice, so it is at early stages, but I found this research one of the most groundbreaking presented AES.
3 – Not all is gene therapy: Many exciting progresses with small molecule drugs
We had a very good year of progresses in small molecule drugs (“normal medicines”) for epilepsy, including for rare and non-rare epilepsies.
Probably the most impressive is the Phase 2 data of XEN1101 in adult patients with focal onset seizures, which is the most common seizure type. There are more than 20 drugs approved for treating focal onset seizures, and trials are done with the new drug added on top of whatever the patients are already taking, so it is very difficult to see impressive efficacy in this oversaturated space. Yet XEN1101 achieved a reduction in seizures of up to 52% at the highest dose. This is rare and exceptionally good.
There were also several updates on late-stage programs presented. We saw post-Phase 3 trial data for fenfluramine in Dravet syndrome, and for ganaxolone in CDKL5 Deficiency Disorder, both looking really good. Ganaxolone has also shown efficacy in PCDH19 epilepsy in a Phase 2 study, with 61,5% of seizure reduction. There is an ongoing trial in young children with diazepam nasal spray as a rescue medication, and Takeda presented how they plan to count seizures in Lennox-Gastaut syndrome in their Phase 3 trial with soticlestat which has a novel glutamatergic mechanism of action.
And I liked to see several innovative compounds coming up in the earlier pipeline. Eisai has a GAT1 inhibitor called E2730 as a potential alternative to tiagabine. A KCC2 activator might have potential for treating seizures in Rett syndrome. Praxis has a sodium channel blocker called PRAX-562 with preclinical efficacy for SCN8A and SCN2A Gain-of-Function epilepsies, and Xenon has sodium channel potentiators with preclinical efficacy for Dravet syndrome (SCN1A).
I might be missing some compounds, but it is clear that 2021 was a very good year for the developments of medications for epilepsy, with some really strong data in clinical trials and in the “real world”, and with new drugs that have novel mechanisms of action at early discovery stages.
4 – Understanding genetics in epilepsy
It is likely that most epilepsies have some genetic contribution. As we learn more about it, and genetic testing becomes more common, we will find many more individuals with rare genetic syndromes and start finding sub-groups of patients with “common” epilepsy that might respond better to certain treatments.
Xenon Pharma has a drug in Phase 3 for KCNQ2-related epilepsy, and they are one of the sponsors of the Invitae genetic testing program for epilepsy. So they interrogated the data from Invitae to see how many of the cases are due to KCNQ2 mutations. I knew this disease was one of the most common genetic epilepsies, but I did not expect the numbers that they got: KCNQ2 is by far the leading cause of neonatal epilepsy, and still the most common in children under 6 months of age. We urgently need early genetic testing for epilepsy to not miss these cases!
There are still new genetic causes of childhood epilepsy being found, like SLC7A3 and SYNJ1 mutations, and scientists are able to create mouse models of genetic epilepsies that require mutations in more than one gene. And a large collaboration of scientists has put together a portal called NDD-CNV to collect information on copy number variations in neurodevelopmental disorders (many with epilepsy) to assist with variant interpretation, which is very important for syndromes like Dup15q and 8p disorders.
And we saw several genotype/phenotype studies where scientists try to see if different types of mutations produce different prognosis which is a common question from parents after genetic diagnosis (see for example SCN1A and STXBP1). There is a little bit of correlation when you compare mutation type in hundreds of patients, but it doesn’t seem that we will be able to predict the development of a child just knowing the specific mutation that they have in a given gene. There was also a massive study in 11,500 patients with epilepsy that relied on phenotype keywords to see if they could be matched to genes, and it identified certain keywords that are preferentially associated with some syndromes like “hemiclonic” and Dravet syndrome, and that suggests that we could potentially use good phenotypic descriptions to prioritize genes to test in these patients. I still believe the best option is to just sequence all people with epilepsy for all epilepsy genes or the entire exome/genome, with particular urgency for young children.
5. Patient groups are building research-enabling platforms
This year we saw some very important research efforts led by patient groups, with the purpose of making it very easy for scientists and companies to work in their disorders:
The NDD-CNV portal is hosted by the Broad Institute and co-developed with the Dup15q Alliance, Project 8p, and Ring 14 US and Ring14 International. It is a natural synergy among structural variant disorders (as opposed to point mutations) and I encourage you to check out the link.
A similar effort, the GRIN Portal, is also hosted by the Broad Institute and developed in partnership with several national and international GRIN patient organizations, and provides information about the genes, the disorders that they cause when mutated and the consequences of specific variants to facilitate interpretation.
Yet another portal hosted by the Broad Institute is the SLC6A1 Portal, with analogous information about SLC6A1 and also with collaboration from patient organizations.
Shout out to Dr Dennis Lal from the Cleveland Clinic and the Broad Institute for making those three portals positive (and more, like the sodium channel portal), and being such an amazing advocate and promotor for patient-led research in genetic epilepsy.
The SYNGAP Research Fund and Ciitizen have also brought together the largest cohort of SYNGAP1-related disorder patients in a platform to collect real world evidence, which will help design clinical trials for this syndrome. And I already mentioned the SCN8A Natural History Study and STXBP1 Natural History Study which are also happening thanks to the effort and leadership of patient organizations.
I believe this type of effort is the best investment that patient groups can do, more than investing on any single research program. It requires that the patient group takes a leadership position in the field and builds the blocks that will facilitate and de-risk individual programs. And it has required a cultural change where clinicians and academic scientists are now very open to partnering with patient organizations and co-leading these efforts. 2021 was a great example of this.
LOOKING INTO 2022
As we look into the new year there are some planned milestones that I look forward to, and some things that I hope will also come true:
I look forward to efficacy data from STK-001 in Dravet syndrome, and the initiation of clinical trials with ETX101.
I look forward to ganaxolone getting approved for CDD, and fenfluramine showing positive results in the ongoing Phase 3 (if interim data is already available by then)
I look forward for more genetic therapies moving into trials. There are several preclinical programs with ASOs and viral-based gene therapies, and in the coming years we should see several of these moving into trials.
And I look forward to seeing progress with several small molecules that target disease-causing proteins, as in KCNQ2, SCN2A, SCN9A and SCN1A Dravet syndrome. Short of gene therapies these might be the best treatments for these disorders.
Ana Mingorance, PhD
Note for parents of someone with Dravet syndrome: when fenfluramine and CBD started trials in Dravet they already had data from adults with other diseases, same for soticlestat and others, so by the time they started trials in Dravet they could immediately administer the drug at the therapeutic dose daily for three months to see efficacy. That is different with STK-001 because it is a disease-specific treatment targeted to people with deficiency in SCN1A so they had to start trials directly in patients and figure out the tolerable and effective dose also in patients, going little by little and starting with low doses. The way I see it, it wouldn’t be correct to compare the current Phase 1/2 Stoke trial with a Phase 3 with other drugs. They are still building up to getting to the best doses, and so far so good.
Disclaimer: These are my own impressions from the presentations and topics that I was most interested in. I write these texts with the parents of individuals with rare epilepsies in mind, so excuse also my lack of technical accuracy in parts.

MAIN LESSONS FROM THE 2021 CDKL5 FORUM
For the past seven years the Loulou Foundation hosts an annual meeting where scientists and drug developers working on CDKL5 deficiency, together with representatives from patient organizations, meet to discuss the latest advances.
Here are the main news and take-home messages from the 2021 CDKL5 Forum that took place in November 1-2 2021.
For the past seven years, the Loulou Foundation has hosted an annual meeting, the CDKL5 Forum, where scientists and drug developers working on CDKL5 Deficiency Disorder (CDD), together with representatives from patient organizations, meet to discuss the latest advances in the field. You can find written summaries from the past three meetings here: 2018, 2019 and 2020.
The 2021 CDKL5 Forum edition took place November 1-2, and for the second year, due to the ongoing pandemic, it was held fully on-line. The Loulou Foundation turned this into an opportunity to also host a series of workshops prior to the 2-day conference, turning the Forum into a giant Think Tank. This made this year’s Forum quite unique, and I hope the pre-conference workshops are here to stay.
I know that many in the CDD patient community are waiting for this written summary, so I will try to extract the main conclusions from the Forum with this audience in mind. But I want to start with a warning: if you are reading this summary mainly looking for an answer to “when are the gene therapy trials for CDD starting?”, the best answer that we have today is “we don’t know yet”. I understand this might feel disappointing to some, as if we are not progressing fast enough, and potentially make you feel a bit hopeless. That’s only human, so I want to acknowledge that upfront.
In the next paragraphs I will walk you through the magnitude of the effort and the tangible progress that has taken place around research and treatments for CDD, and that was presented at the Forum. I invite you to read it in the spirit of celebrating each step and each progress and small victory along the way, because we have come a very long way, and this year the progress in biology understanding and therapy development has been particularly significant.
It is remarkable how far we’ve come in the last few years, and the size and commitment of the community of scientists and companies that has grown around CDD.
Because of that, I have structured the summary of this year’s Forum around the following themes: hope, tangible progresses, strategy and partnerships.
1. HOPE
The 2021 Forum started with the voice of the patient. This year it was IFCR’s President Karen Utley, who shared with the audience the life of her daughter Samantha, and the impact that CDD has on Samantha and her entire family. Samantha sees 14 different specialists a year and takes 15 different medications, which gives us some quantitative data for the complexity of this disorder. From the many great messages from Karen, I keep two: (i) small improvements can be life changing for patients and families, so please don’t focus only on cures and keep working to make more smiles and happy days, and to (ii) please keep hope alive for those who are feeling too overwhelmed to stay hopeful.
Her plead to keep hope alive was adopted as a motto by many of the conference speakers in the two days that followed.
There were several moments during the conference where science showed us there are many reasons for hope. These are some of those reasons:
Hope because mice show us that CDD is a good candidate for genetic rescue

Just two weeks before the Forum we celebrated the publication of a landmark study showing that CDD is reversible in mice (article link). Zaolan (Joe) Zhou, from the University of Pennsylvania, also presented the results of his study at the Forum, which show that CDKL5 is needed throughout life to keep proper neuronal functioning and that, in mice, turning “on” the CDKL5 gene in near-adult mice that grew up with CDKL5 Deficiency leads to large reversal of most symptoms. This is very valuable because it helps us learn what is biologically possible, and that CDD is not just a disease of neurodevelopment but also of neuronal maintenance.
In layman terms, it appears that in CDKL5 Deficiency, the brain developed fine and the problem is that neurons are not talking to each other correctly. So once we put the protein (or the gene) back they can start talking to each other correctly, because nothing was really lost: there was no neuronal death or failure to form those neurons, and they are not in the wrong location, which are all things that happen in other neurodevelopmental diseases. This study is a big reason for hope, and Joe received the CDKL5 Forum Award for Lab of the Year for this contribution to the field.
Hope because other diseases are already seeing great results
As part of the gene therapy session, Krystof Bankiewicz, from Ohio State University, showed us videos of children with AADC Deficiency who participated in a clinical trial for a gene therapy to deliver to their brains the AADC enzyme that they are missing. If you saw the videos you probably had chills. Patients with AADC Deficiency have very strong hypotonia, intellectual disability, dysautonomia and other complications due to missing an important enzyme necessary for producing dopamine and serotonin. Prior to treatment, the mobility of these patients is so impaired that they reminded me to several of the boys with CDD, unable to even hold their heads up. In the videos, we saw children go from that extreme hypotonia state to seeing changes 1 month after gene therapy, sitting after 6 months, walking with assistance of DAFOs after a year, and totally unrecognizable walking by themselves after 18 months. Krystof explained that many of the patients also became verbal, which was highly unexpected. While we don’t know yet how fast and how much people with CDD will improve once we have a gene therapy for them, seeing the results in this other enzyme deficiency help us learn what is biologically possible, and it is a big reason for hope.
Hope because science (and scientists!) keep coming up with great genetic tricks very quickly
While we are today very focused on gene therapies to bring an extra copy of CDKL5 to each neuron in the brain, we got to see at the Forum some of the science that is coming afterwards, and that will allow us to fix the mutated gene in patients with genetic diseases. David Liu, from the Broad Institute, showed us how he is using an advanced version of the CRISPR system to correct mutations where one letter is missing, and showed us how they can do it already in cultured cells from a girl with CDD (this is still all very early and being optimized). And Feng Zhang, also from the Broad Institute, showed us another advanced version of the CRISPR system that instead of correcting one letter in the DNA can swap large sections of a gene or an entire gene. So we can imagine one day getting a single gene therapy that gets to the brain and swaps the entire CDKL5 gene for a good copy, and that could potentially be used for kids with all types of mutations.
But before we get to see these gene editing approaches turned into actual treatments, we have many other therapies coming our away. I review those in our next section: tangible progress.
2. TANGIBLE PROGRESS
Progress in three gene therapies
Last year at the 2020 Forum we talked about the two gene therapies for CDD that had shown to have efficacy in mice deficient in CDKL5, or as we call it in science had “preclinical Proof-of-Concept”. We also talked about how gene therapies need to go through three stages: first prove that you can treat a mouse, then prove that you can use it in larger brains (non-human primates) since it is much more difficult than in small mouse brains, and then finally start the first trials in a very small group of patients to determine safety before exposing more patients.
This year we saw not two, but three gene therapies, and how the most advanced ones are now working on that second stage that goes between showing that you can treat mice and being able to start clinical trials.
The gene therapy that we saw presented for the first time is the program between the University of Pennsylvania (Penn) and the company Amicus, and it is a very interesting hybrid between gene therapy and protein (enzyme replacement) therapy. They call the approach cross-correction, because the therapy uses a virus to bring to neurons a copy of the CDKL5 gene (this is a gene therapy), with a special version of CDKL5 that will produce a secreted protein. The idea is that the neurons that are successfully corrected will act as local sources of enzyme to supply to neighboring cells. This was the first time that we saw mouse data from this project at the Forum, and the researchers are trying to see how much more efficacy they can get by using this special version of CDKL5 as opposed to plain CDKL5.
For the other two gene therapies, from Penn (in collaboration with Elaaj Bio) and Ultragenyx, we got to see some of the work that goes into getting the therapies ready for patients now that we know both of them work in mice.
The gene therapy program from Penn/Elaaj came with new EEG data, showing that it is not only the symptoms but also the EEG abnormalities in mice with CDKL5 deficiency that get corrected with the gene therapy. And more importantly, they showed two newer versions of the gene therapy, more advanced than the one we saw last year, and how with each iteration they are getting better and better distribution in the non-human primate brains. They are making these upgrades trying to get as much expression of CDKL5 and as broad as possible, and as the presenter said, “we are getting closer to the target of robust expression in the brain” that they want to see before moving into clinical trials.
The team from Ultragenyx also dedicated most of their presentation to talk about those next steps after seeing efficacy in mice with CDD (which we saw last year). They told us about the need to fully evaluate safety and toxicology in animals before entering the clinic, and also about all of the additional work that they are doing to prepare for clinical trials involving natural history and biomarker data. This was very impressive, and showed how Ultragenyx has many people in the clinical and regulatory teams and many other teams in addition to their preclinical researchers all working on the CDKL5 program.
So we don’t know yet when trials might start, but we know that these three programs keep getting closer and closer to the clinic, and that we have some of the best people in medicine working on solving all of the difficult barriers that need to be overcome to take these treatments into clinical trials (like better brain distribution, and development of biomarkers, and clarity on clinical scales to use). You will see more of this in the last section about strategy and partnerships.
Progress with ganaxolone
If everything goes well, ganaxolone will become the first drug to be approved for the treatment of CDD. The FDA plans to make a decision in March 2022, and the EMA some months later. In the meantime, Marinus has an Expanded Access Program ongoing which is now also open to Europe.
What I found really exciting from the Marinus presentation was data from their open-label studies, where they showed how patients from the Phase 3 trials are doing as they rolled over into the open-label extension. Since it is also openly available in their website and not confidential, I am going to copy their slide here because the data looks really good and it is better seen in the graph:

In the solid green bars, you see the patients that received ganaxolone during the Phase 3 study, and then continued taking it in the open-label extension. Remember that the study is blind, so no one knows if they are taking placebo or ganaxolone during the 17-week study. These patients had a seizure reduction of 30% at the end of the trial, and for the following 12 months their seizure control got better, staying more around 40-45% seizure reduction. This is not trivial, because it is common for patients with CDD to respond to a new medication for 3 or 4 months and then lose efficacy, while ganaxolone doesn’t show signs of losing any efficacy after a year, all the contrary.
And in the striped green bars you see the patients that received placebo during the Phase 3 study, but then started taking it in the open-label extension. Because the study is blind no one knew during the 17-week study if they were getting the drug and not responding to it or if they were in placebo. On average this group had seizure reduction of only 6,9% during the trial (since they were not on the drug), and as they rolled over into the open-label extension their seizure control got better and better until they caught up with the ones that we already taking ganaxolone in the trial.
This is a beautiful graph, that shows that ganaxolone has better efficacy in CDD than what is seen only during the double-blind phase, and that you have to give it enough time to show its full potential. It is also quite encouraging to see how stable the efficacy of ganaxolone remains over time.
Progress with fenfluramine
Fenfluramine is an anti-seizure drug that is already approved for Dravet syndrome where it has exceptionally high efficacy in reducing seizures. After a successful small pilot study in CDD run at NYU, the company Zogenix has been working on preparing for a Phase 3 trial of fenfluramine in CDD.

And the Zogenix team gave us the great news at the Forum that the Phase 3 trial with fenfluramine in CDD has already started and is already enrolling patients in the US clinical sites! They are enrolling children and adults with CDD as young as 2 years old, and once they have enough safety data they might open it to 1-year-olds and older, and patients need to have active epilepsy since that is what the study is measuring. For the trial, Zogenix is looking for 100 participants.
This is really good news because it means that at least 100 patients with CDD will get to try fenfluramine, and if it has in them even a fraction of the efficacy that we have seen in Dravet syndrome and in the small study in CDD, it will be very meaningful for those patients and their families.
3. STRATEGY AND PARTNERSHIPS
The Forum becomes a Think Tank
During the week prior to the conference, the Loulou Foundation hosted focused workshops that covered four areas of priority for the development of therapies for CDD:
Spontaneous and induced seizure activity in CDKL5 knockout animal models
New technologies for improved therapeutic cargo delivery in the central nervous system
Biobanking of patient samples and fluid biomarker discovery/validation to support CDD therapeutic development
Functional biomarker development/validation and inclusion in clinical development plans
For each workshop, the Foundation invited a group of academic, industry and clinical researchers, and for some also patient representatives, to try to map out (i) where we are, (ii) where we need to be, (iii) what are the technologies or fields that we should be paying attention to, and (iv) how we can get organized to address those challenges including specific proposals for collaborations. This is what turned the 2021 Forum edition into a true Think Tank, and the outcomes from each of the workshops were presented during the main conference to share with the entire community.
To give you an example for how these workshops worked, when talking about new technologies for delivering genes to the brain, we covered all approaches from engineered viruses to synthetic biology, nanoparticles and the possibility to attach CDKL5 to proteins that cross the blood-brain-barrier to use them as shuttles. And we had scientists from companies that are developing all of these approaches sit on the same workshop and share with everyone else their thoughts and proposals. This level of collaboration among traditional competitors is highly unusual, and it is one of the themes that we kept seeing during the conference: the building of a scientific community around CDD that includes many companies joining forces to help us get to better treatments. You will hear more of this later, when I talk about the CANDID study.
The long road to solving the translational puzzle
Translational medicine refers to the bridge between treating mice and treating people. It seeks to answer questions such as: “how will we know that we have increased CDKL5 protein in the brain?”, “how will we know if we are improving synaptic function?” and “how are we going to measure improvements in patients in the trial?”.
To answer these questions, we need to develop ways to see things, which can be biomarkers (like things you can see in a blood test, or brain scan) or clinical scales. Because CDD is a relatively newly-described disease, we still don’t have these biomarkers and scales.
The good news is that there are many collaborative efforts ongoing to address all these questions and develop all these biomarkers and outcome measures. This was a central theme of this year’s Forum and the subject of two of the pre-meeting workshops, with important presentations on these topics:
Need and plans for a biobank of patient samples, with standardization of analytical techniques and data sharing including universities and companies
EEG-based biomarkers, presented by Eric Marsh from CHOP, as a way to measure dysfunction of brain activity in mice and patients with CDD, to detect potential restoration with treatments
Blood-based biomarkers, presented by Max Bianchi, from the Trinity College Dublin, as another way to measure dysfunction of brain activity in mice and patients with CDD, to detect potential restoration with treatments
A US and Australia NIH-funded study coordinated by Tim Benke from the University of Colorado to also test and validate biomarkers as well as new clinical scales for CDD in an observational study
An international observational study coordinated by the Loulou Foundation in partnership with 7 pharma companies to test and validate clinical scales for CDD, called the CANDID study (see also next section)
In addition to helping us solve the translational puzzle, what all of these efforts and studies have in common is that they all require the active participation of the patient community. This could look like organizing the collection of some blood samples at your next national families meeting to then send them to Dublin, or participating in an EEG study in Philadelphia or Boston, or volunteering to participate in the observational studies that will take place in clinics from around the world. You will likely hear of some study near you!
So if you have a family member with CDD, I encourage you to consider participating in some of the studies as well as enrolling into the CDKL5 Registry (this one only needs data, no samples or visits to a clinic needed).
We cannot answer all of these key translational questions without you.
A first-of-its-kind partnership
Just a week before the Forum, the Loulou Foundation announced the launch of a pre-competitive industry collaboration involving seven biopharmaceutical companies together with the Loulou Foundation. The consortium will run an observational study specifically designed to evaluate the feasibility and suitability of a collection of clinical outcome measures in people with CDD from all ages.
Xavier Liogier, from the Loulou Foundation, explained how this study fits into a larger roadmap for preparing the CDD field for the arrival of better therapies and cures, and that already started with the PFDD meeting in 2019 and getting an ICD-10 code for CDD.
Observational studies are like a clinical trial but instead of testing a drug you are testing the clinical scales to check that they are good for that disease. These studies are very important to be ready for clinical trials that look at several symptoms or disease domains. Usually, when companies run observational studies of this kind, they do it alone, so in diseases where there are many companies developing therapies we might end up seeing several observational studies, each one run by a different company with a slightly different design.
We wanted to avoid this for CDD.
The idea for the CANDID study was born two years ago as a big hairy audacious goal: what if we could get all of the companies working on CDD come together, and co-design a single observational study that would meet everyone’s needs, get also the FDA to review it while we are at it, and then run that single study with all of the companies working together to minimize the number of observational studies that we need in CDD.
It sounded good. And all of the clinical teams at the different companies that we talked to also told us that it would be something they would support. That’s what gave us the conviction that we needed to try to make it happen. The problem was that we had no reference for other collaborations of this type. There was no book that we could read to know how to do this. This was going to be a first-of-its-kind partnership.
So for the past two years we had to figure out a way to co-design a study among so many parties without the study dying of decision paralysis, and we had to figure out the best way to build a consortium to govern the study where all companies would be equal partners. We also had to grow as a Foundation to be able to lead and coordinate these efforts, which brought us new team members like Xavier Liogier, who has the ideal human and professional mix to lead this study, and new strategic partners in clinical operations to help run this observational study with the very same standards as a pharma-run Phase 3 trial.

This is why I am so proud to be part of this story, as part of the Loulou Foundation team that made the CANDID study possible. We knew that all the companies working on CDD would like to see a consortium-run study, and we all understood that we needed to try to avoid having multiple corporate observational studies in CDD. So we figured out how to make this first-of-its-kind partnership happen, and in the process, wrote the first chapter of that book.
The CANDID study is about to start and will look for 100 participants of all ages (with active epilepsy or not), hoping to include study sites in the US, Canada, UK, France, Spain, Italy, Germany and Russia. This will help us make the study accessible to the large majority of patients diagnosed with CDD.
Strategy and partnership also on the advocacy side
Natalie Ladly, secretary for the International CDKL5 Alliance, helped close the conference with a presentation about the Alliance and a call for action.
There are 17 patient organizations under the Alliance (check them out at https://cdkl5alliance.org/) and one of the top priorities is to help the community be trial-ready. Natalie explained that they have 47 clinical trial sites around the world, at least one on each of the countries under the Alliance, and made a call to the industry and the medical community to reach out to the national patient groups and the Alliance and tell them what would help them (industry and clinicians) the most. Essentially, “help us help you”, and “by the way we have solved your problem for finding trial sites”.
I believe every year I write a longer summary. There is just so much going on, and so many news and progresses in many areas, that it is difficult to know what to cut from the summary. My main impressions from the 2021 CDKL5 Forum are that we are seeing solid tangible progresses in many areas, and that we seem to be turning a corner, where the ratio of individual labs working on CDD versus partnerships is changing and now a majority is working on the same agenda to solve the same problems together. This partnership model was started by the academic and clinical centers, and it has now also reached biotech and pharma companies.
This is why I want to commend in particular the professionals from so many biotech and pharma companies (we had 24 companies at the Forum!) that came to the Forum and sat down with their traditional competitors to share their best ideas for CDD and agree on joint efforts to make these happen. And I believe that the industry consortium behind the CANDID study is a testimony to that spirit of breaking previous barriers to industry collaboration, not just by saying nice words, but by working together to make progress actually happen.
And that is why hope, tangible progresses, strategy, and partnerships, where the themes of the 2021 CDKL5 Forum.
I hope you enjoyed this summary. Please let me know your thoughts in the comments.
Ana Mingorance, PhD
Disclaimer: These are my own impressions from the presentations and topics that I was most interested in as a scientist and patient advocate, and not an official text about the Forum by the Loulou Foundation. I write these texts with the parents of individuals with CDD in mind, so excuse also my lack of technical accuracy in parts.

CDKL5 DEFICIENCY IS REVERSIBLE
CDKL5 deficiency is biologically reversible, bringing much hope and creating a sense of urgency for the development of restorative treatments for CDKL5 deficiency disorder (CDD). These are the conclusions of a publication from Zhaoland (Joe) Zhou’s lab from the University of Pennsylvania. The study was just published in the Journal of Clinical Investigation, and it is poised to become a landmark study in the field.
CDKL5 deficiency is biologically reversible, bringing much hope and creating a sense of urgency for the development of restorative treatments for CDKL5 deficiency disorder (CDD).

These are the conclusions of a publication from Zhaolan (Joe) Zhou’s lab from the University of Pennsylvania. The study was just published in the Journal of Clinical Investigation, and it is poised to become a landmark study in the field.
It has been almost 15 years since a similar publication from Adrian Bird in mice with Rett syndrome due to mutations in MECP2. Bird’s lab showed that Rett syndrome was reversible if the gene was returned to adult mice who had developed without MECP2. The study told us that it is likely never too late to rescue MECP2 deficiency, and it encouraged research into the genetic rescue of Rett syndrome in patients. The experiment (and the investigator) became so famous, that gene therapy researchers from universities and industry have been asking for the last 15 years “has anyone already done the Adrian Bird experiment for this gene?” for any genetic syndrome that they consider working on.
Joe Zhou and his colleagues just did that for CDKL5. And the response is affirmative: CDKL5 deficiency is biologically reversible.
Here is a summary of these findings, and what they mean in the context of developing curative treatments for CDD. There is also a press release from Penn here.
WHAT THE PUBLICATION SHOWS, IN LAYMAN’S TERMS
I love the elegance of this study, and I am impressed by the amount of work and exquisite diligence that went into creating this publication. This is why I feel so strongly about the importance of these results.
Terzic et al (2021) creates two types of genetically modified mice. One type is mice that are born with normal CDKL5 expression, but that can stop expressing CDKL5 when the investigator decides. The other type are born with CDKL5 deficiency, but can produce CDKL5 when the investigator decides. Essentially there is one mouse strain where scientists can decide when the disease will start, and one where scientists can decide when it will stop. And they use these mice to answer key questions about the biology behind CDKL5 deficiency.
Because the protein CDKL5 is expressed in the brain throughout life, the scientists hypothesized that perhaps it is important also throughout life, not just in early development. This is not trivial. People born with a bad copy of CDKL5 develop a disease called CDKL5 deficiency disorder (CDD) that starts very very early in life, leading to global developmental delay in addition to seizures and other complications. Because it starts so early, it looks like having enough CDKL5 is important for very early development, and maybe if you make it past all those critical early stages of post-natal development with normal expression of CDKL5 then you will always be ok even if you stop producing enough CDKL5 later in life. Short answer: you are not.
It is never too late to develop CDD. That is why the protein CDKL5 is expressed in the brain throughout life, because it is always needed.

This is what the publication shows, by letting mice develop normally and then turning off their CDKL5 gene when they are young adults. By that age, mice that are born with CDKL5 deficiency already have all their symptoms, which include cognitive, motor and behavioral deficits, as well as alterations of neuronal excitability and synapses. Same as for people with CDD. And the first important finding of the publication is that if you let mice grow up to that age completely normal, and then turn off their CDKL5 gene, they will progressively develop almost all the same symptoms of the disease (except for less motor symptoms). Pretty much as if they had been born with CDKL5 deficiency.
And that is the first conclusion: CDKL5 deficiency looks like a disease of neuronal maintenance, not so much of neuronal development. We talked about that at the 2020 CDKL5 Forum (read it here).
The second part of the study gets better: if CDKL5 is so important for the mature brain, what happens if we bring it back at that point? How much can we get back?
For this the Penn team uses the second type of mice, the ones in which they can stop CDKL5 deficiency at any age they want. And they let them grow through all those key developmental ages with CDKL5 deficiency, and they let them display all of the disease symptoms. And then, when mice are early adults, they turned on the gene so they no longer lacked CDKL5. This is the mouse version of what would happen if we already had the perfect gene therapy for CDD and we treated patients that are no longer children.
And the results are beautiful and make this study a landmark in the development of curative treatments for CDD. Barbara Terzic and Joe and their colleagues show how later-age restoration of CDKL5 expression rescues most CDD deficits. They re-start CDKL5 expression in mice that are of young adult age, that already have developed all their CDD symptoms, and then see reduction or full rescue in many motor, cognitive and behavioral readouts. Mice with CDD don’t develop easy-to-measure seizures, which is a difference with how the disease presents in people, but the Penn group was able to interrogate brain circuit hyperexcitability using an excitatory neurotransmitter similar to glutamate to “poke” neurons, and saw how the hyperexcitability that comes with CDKL5 deficiency was also rescued by reintroduction of CDKL5 expression. They also used this neurotransmitter to induce seizures in the CDKL5 deficient mice since they are prone to epilepsy, and saw how this was also rescued by reintroduction of CDKL5 expression. They even looked at the presence of receptors in synapses, which is altered by CDKL5 deficiency, and this was also restored in the adult-rescued mice. So I want to highlight again how thorough and well-done this study is: it is very clear from this study that reintroducing CDKL5 expression later in life is able to correct functional and behavioral deficits across the board, not just a few symptoms.
And that is the big beautiful conclusion of the study: the promise of disease reversal in CDD, across a broader-than-expected time window.
WAS THIS NOT OBVIOUS OR KNOWN BEFORE?
No it wasn’t.

During brain development neurons need to be born, then need to migrate to where they are needed in the brain, grow connections to the parts of the brain that they need to be connected to, and then do their job properly remaining in place until we get old and neurons start dying. In many neurodevelopmental diseases neurons might fail to develop properly, or might migrate to the wrong places, or not connect with who they should, or suffer from neuronal loss. And for all those it is too late to come with the gene after those problems have happened.
We knew that CDD looked quite good for a potential genetic rescue in older individuals. We knew the neurons where there, in the right location, that brain connections looked good, and there were no signs of neurodegeneration. At the biological level CDD looks more like a channelopathy, like Dravet syndrome (Nav1.1 channel), than a proper developmental disease.
But from thinking “the odds are good” to being able to say “at least in mice this disease is reversible” there is a long stretch. That is why this study is such a landmark.
The results from the Adrian Bird study in Rett syndrome are actually an exception. Similar studies in related diseases have failed to show such an across-the-board rescue. For example in Phelan-McDermid mice, reintroduction of the SHANK3 gene in adult mice only rescues some symptoms, and early postnatal intervention is needed for across-the-board rescue (link). And in the Angelman mice (Ube3a gene), you can only rescue many of the symptoms if you restore the gene during early development, and the authors concluded that “adult reactivation of Ube3a is only minimally efficacious as a therapeutic intervention” (link).
So with CDD starting so early in patients, and with the clinical symptoms being so severe, it was not obvious at all that CDD would look so promising for rescue in older individuals and not more similar to the mouse study for Angelman.
WHAT IT MEANS FOR GENE THERAPIES
This study means that we now know that CDKL5 deficiency is biologically reversible. Now we need to get the tools to do that in the clinic.
What these genetic rescue studies in mice have in common is that they use mice that are genetically re-programmable, where we as scientists can choose to turn on or off a particular gene defect. People is not born like that, we cannot choose to turn off their genetic disease. We need therapies for that. These could be therapies like gene therapy, where we bring a new copy of the CDKL5 gene to their neurons, or other therapies like gene editing that fixes the mutation that they have, or even protein therapies if we can supply enough CDKL5 protein to their brain.
So there are many technical possibilities to restore CDKL5 in patients, but there are also many challenges to do this. This is why developing this type of treatment takes so long. One of the main challenges is getting to all (or most) neurons in the brain. CDD is not a disorder that impacts a small brain region, and where we only need to fix it there with an injection. It actually involves the entire brain, and we would need to make sure all (or many) of those neurons have sufficient CDKL5 as a result of a treatment. This sounds simple but is tremendously difficult. Understanding how many neurons need to be rescued to have enough efficacy, and how much CDKL5 exactly do they need (is it ok with half of the amount?), and having the right therapy that can do that in a safe way… it is not easy.
But the good news is that this is a challenge for ALL neurological diseases, so scientists and companies around the world are trying to make sure we know how to make treatments that can put back a gene (or fix it) throughout the brain. This will benefit CDD and many others. And it is also good news that we already have several of these scientists and companies working on developing these treatments for CDD, and we have already seen some early results with two of these gene therapies in mice with CDD (see here).
It will only get better from here. I expect this study to attract more attention, more research and more resources, to developing curative treatments for CDD now that we know biology is on our side.
In summary:
The part of the puzzle that we could do nothing about, whether the brain has a narrow temporal window where it needs CDKL5 or not, is the one where we got very lucky. CDKL5 deficiency is biologically reversible.
The second part of the puzzle is to build those therapies, and that one is not about luck, but about science and time and money. We can do much about it.
This is like the difficulty of building a rocket to go to the moon: it is hard, but you know that your destination is waiting at the other side.
Now that we know that moon exists, that the disease is biologically reversible, all hands on deck to build that rocket.
PS1: Thank you to the Penn teams for working so many years in this study. This study had financial support from the Loulou Foundation, IFCR and several NIH grants. Science is not easy or cheap. And thank you for making the publication open access!
PS2: The 2021 edition of the CDKL5 Forum is coming up in November 1-2, and this study is only one of the several good news that will be presented at the meeting.
Ana Mingorance, PhD
2021 CDKL5 ALLIANCE Virtual Family Conference
Last weekend, the CDKL5 deficiency disorder (CDD) community gathered in front of their computers for a virtual annual meeting organized by the International CDKL5 Alliance. For the 2021 Alliance meeting, the organizing team commission a series of pre-recorded videos to a large range of speakers and published the videos with subtitles so that all CDD families from all around the world could watch them at the same time. You can also access the videos at the CDKL5 Alliance website. Here is a personal summary of the 2021 CDKL5 Alliance meeting.
I miss conferences in person a lot. And the type of conference in person that I miss the most is the annual patient conferences organized by the rare epilepsy patient organizations that I collaborate with.
Last weekend, the CDKL5 deficiency disorder (CDD) community gathered in front of their computers for a virtual annual meeting organized by the International CDKL5 Alliance. Did you know that there are about 20 countries in the Alliance? That is how large and connected this community is. The last face-to-face meeting took place two years ago and I strongly encourage you to read the summary from that meeting.
For the 2021 Alliance meeting, the organizing team commissioned a series of pre-recorded videos to a large range of speakers and then published the videos with subtitles so that all CDD families from all around the world could watch them at the same time. You can also access the videos at the CDKL5 Alliance website.
Some parents of children with CDD have asked me to write a summary of the virtual conference, since a written summary is always easier to translate and digest. For all of you here is a personal summary of the 2021 CDKL5 Alliance meeting, sprinkled with some opinions as I often do.
1. THE SCIENCE: HIGHWAYS AND HEADQUARTERS
Prof John Rouse from Dundee reviewed where we are in our understanding of what CDKL5 does. In the recent years we have made large progresses on our understanding of what CDKL5 does in the cell and therefore what goes wrong when CDKL5 is missing.
Today we know that the protein CDKL5 turns other proteins on and off as if they had a light switch, and many of these proteins that are controlled by CDKL5 are associated with the neuronal skeleton (the cytoskeleton). The neuronal skeleton is not like our bones, which are essentially there to give us some structure. In cells, the skeleton is more like highways that are used to transport things to where they need to be, for example making sure that glutamate receptors get to the right spot in the surface of the neuron for the neuron-to-neuron connection to work fine.

And in addition to being important for the highways, it turns out that CDKL5 has a second job at the cell nucleus, which is basically the headquarters or control center in the cell from where all decisions are made. But what could CDK5 be doing there? This is what keeps John awake at night, so he has put together a team of scientists to try to answer this question. It turns out that in the nucleus, where all of the copies of all of our genes are located, the genes (made up of DNA) are constantly suffering small damages that the cell quickly repairs so that all genes work properly. And CDKL5 is a member of that DNA damage surveillance team. It does that by turning on other proteins that are needed to do the repair. And it also helps the cell read some of these genes. So CDKL5 is really important for the nucleus to function properly.
With the very complex (and multiple!) roles of CDKL5 in the cell, it is clear to me that the best way to fix the disease is to put the gene or the protein back, or to fix the mutation. Anything that will produce functional CDKL5 protein again. Trying to bypass a protein that is missing is doable if the function was only one, but for CDKL5 there are too many functions to try to fix, so I would focus on bringing CDKL5 back.
2. CLINICAL TRIALS: WHERE ARE WE TODAY
Before a drug or a gene therapy can be approved to be used to treat patients it has to progress through clinical trials. I think this year with the coronavirus vaccines we are all very familiar with how clinical trials work, so I will only explain that in rare diseases like CDD we have two stages of clinical trials: Phase 2 trials, where a small group of patients get the treatment, and Phase 3 trials, where many more patients from many countries get the treatment and usually half of them get placebo (but are offered the treatment at the end of the study anyways). If the Phase 3 is successful, then the company can apply for marketing authorisation. The entire process for trials and regulatory review for approval ends up taking 4 to 5 years.
We still don’t have gene or protein therapies for CDD, so while we wait for scientists to develop them we are trying to discover medicines that could treat the symptoms of CDD, helping all patients have a better life until we get to the curative treatments. We have had some clinical trials already with symptomatic treatments in CDD.
Last year we had very good news about clinical trials: the first Phase 3 trial in CDD, ganaxolone from Marinus, had been successful.
This year we also have very good news about clinical trials: we are about to start our second ever Phase 3 clinical trial in CDD, this time the drug is fenfluramine from Zogenix.
At the Alliance conference, Marinus reviewed their good data with ganaxolone in CDD. They are offering ganaxolone to all of the trial participants that want to keep taking it, and in the USA they are also offering it to other families who want to try ganaxolone before it gets approved. This is done through an Expanded Access Program. Ganaxolone works by acting on GABA, the inhibitory signal in the brain. If everything goes well ganaxolone will be the first drug to be approved for CDD.
Last year there were two Phase 2 studies in CDD. One was with the drug soticlestat, from Ovid and Takeda. This drug works by acting on the glutamate system, which is the excitatory signal in the brain. The companies were testing the drug in four different epilepsy syndromes, and after reviewing their data Takeda has announced that they will run Phase 3 studies in Dravet syndrome and Lennox-Gastaut syndrome. They still have not announced what they will do with CDD, and they did not present at the conference so we will have to keep an eye on any news about this program.
The second Phase 2 study from last year was with fenfluramine, which works by acting on the serotonin system, a modulatory signal in the brain. Fenfluramine has already been approved in the US and Europe for the treatment of Dravet syndrome, where it has impressive efficacy to reduce seizure frequency. Zogenix presented an overall introduction to the company at the Alliance meeting and didn’t talk much about their plans for CDD, but they have already announced that they are starting a Phase 3 trial in CDD with fenfluramine starting this year. Phase 3 trials are international, happening in many countries, so I look forward to the announcement of which countries will be included in this study. And if the Phase 3 results look like the small Phase 2 study results, we could have potentially a second drug approved for the symptomatic management of CDD.
This progression of clinical trials for CDD is exceptional! Only 5% of rare diseases have any drug approved, which means that 95% have nothing. And we are having these two Phase 3 trials back to back in CDD, so we are on track to join that 5%!
But these symptomatic treatments are only the tip of the iceberg. Right below we have a growing pipeline of therapies in development that will correct the lack of CDKL5.

3. THE FUTURE: FROM SYMPTOMS TO CURES
If you have a child with CDD the main answer that you probably wanted to get from this conference is WHEN are gene therapies starting clinical trials.
And I am sorry to say that the answer is “we don’t know yet”. Because we cannot know yet, nobody does. There were four different groups that presented data at the Alliance meeting showing their progresses towards developing a gene therapy for CDD. That is four different shots on goal, four independent efforts like when many companies all started developing vaccines for COVID without knowing how many would succeed and how fast it would happen. And we are following the same approach in CDD: many efforts all at the same time going as fast as they can.
The university of Bologna is developing a mix between an enzyme replacement therapy and a gene therapy. An enzyme replacement therapy is “protein therapy”, a type of treatment where you add the brain the protein that is missing, in our case the protein CDKL5 already made. For a gene therapy scientists use a virus to bring to cells a copy of the CDKL5. It is similar to the AstraZeneca or the Janssen vaccine but instead of using a virus to carry to the body the sequence of the coronavirus spike we use them to carry the sequence for the CDKL5 gene. The university of Bologna is using a virus to deliver to neurons the sequence for the CDKL5 gene, but it has been tweaked so that the protein CDKL5 will then be secreted by those neurons to all the surrounding ones, doing “local enzyme replacement therapy” on top of gene therapy. This is also called “cross-correction”. It is a cool idea. Amicus is doing something very very similar, in addition to classical enzyme replacement therapy with the purified protein. And then Ultragenyx and the University of Pennsylvania are doing the classical gene therapy using a virus to carry the sequence for the CDKL5 gene to the neurons in CDD. That means that we got to hear about FIVE different gene therapy or enzyme replacement approaches at the patient conference, by four groups since Amicus has two different programs. That is a lot.
The university of Bologna showed data using their cross-correction therapy in laboratory cells and mice. This is still early work at a university so it is too early to know how much work the experimental treatment still needs before it could move to trials.
Amicus presented some data about their cross-correction therapy program and their classical enzyme replacement therapy program. It was not an in-depth project review, but more of an overview of all the work that Amicus is doing in CDD which includes not only these two programs but also helping understand better how CDKL5 works and developing biomarkers (detection of special signals using EEG) that will help check if therapies are working in CDD mice and patients much faster. CDD is clearly important to Amicus. They are doing many experiments in mice with both treatments to see how well they could prevent or reverse the symptom of the disease in mice with CDKL5 deficiency.
The University of Pennsylvania presented a lot of data in mice with CDKL5 deficiency using a gene therapy to show that it is possible to prevent most of the symptoms in CDD mice. I say “prevent” because they give the gene therapy to mice when they are babies. I really liked the presentation, because they showed how the gene therapy was effective in many disease symptoms. That is the main different with symptomatic treatments: with gene therapy, you put the gene back and you are treating all of the different symptoms at once by removing the cause of all of them. This group has also run initial experiments in larger animals (macaque monkeys) to check safety and because we need to check that the gene therapy can get to enough neurons in larger brains, which is literally a much larger challenge than delivering the gene therapy to the brain of a mouse which is the size of your thumb nail. They explained that they are currently optimizing the gene therapy so that then they can go to trials.
The last presentation was from Ultragenyx, who are also developing a classical gene therapy similar to the one from the University of Pennsylvania. I also liked this presentation very much because they treated CDD mice at older ages, the equivalent of children 4-11 years of age, so what they showed to us was that it is possible to reverse many of the symptoms in CDD mice. They saw reduction of cognitive, motor and behavioral symptoms in the mice, and even used the gene therapy to correct neuronal connection problems in “human neurons” which were produced from patient skin biopsies and turned into neurons in the lab. Just like for the University of Pennsylvania, they explained that they are currently optimizing the gene therapy to be able to get into many neurons in larger brains (monkey) so that then they can go to trials.
As soon as one of the groups considers that their gene therapy is optimized enough, they will start the process to request a clinical trial permission and at that point we will finally get a date for when clinical trials might start. I hope you all realize how much work all of this is, and how lucky we are to have these five protein and gene therapy programs being all developed at the same time for CDD!
4. DATA, DATA, DATA
There were several presentations about why the international CDKL5 patient registry is so important, the international CDKL5 patient database from Australia, a study to validate several severity and symptom assessment for CDD, and an international observational study that is about to start and that is coordinated by us at the Loulou Foundation.
At the core of all of these efforts is a common theme: we need to understand better the symptoms of CDD and how they change over time (the natural history), we need to identify good scales to measure these symptoms, and this information and scales are absolutely essential to be able to tell if a treatment is working beyond just reducing seizures.
For the registry and the database, you can provide data about your child from your living room, and for the observational study we will try to get as close to your town and living room as possible!
By the way, have you noticed that clinical trials all have names? For example the trial with ganaxolone in CDD is the Marigold study, and the famous trials with spinraza in SMA was called Nurture. The Loulou Foundation is requesting suggestions from the community to name the international observational study so please reach out with your proposed names! (you can get some ideas here)
5. THE COMMUNITY
Although we didn’t get to meet in person, we did get to see many of the visible faces of the CDD patient community at this virtual conference.
Patric Benz from the Swiss CDKL5 patient group was the meeting host on behalf of the German-speaking national patient associations who were the organizers. His greeting in several languages and the fact that the videos were available with multiple language subtitles was a testimony to the reality of rare disease patient communities: at the same time dispersed and united.
During the conference we also saw images of people with CDD kayaking, skiing, ice skating and cycling. If there is something notable about this community it also their fearlessness and determination.
To conclude the conference, Nathalie Ladly from CDKL5 Canada and Antonino Caridi from CDKL5 Italy spoke about some of the big efforts of the Alliance in this last year, like developing the organization manifesto and sourcing a list of all possible clinical trial sites world-wide recommended by the community so that we can accelerate clinical trials. The goal of the Alliance is to expedite treatments and a cure for CDKL5, so of course a global pandemic would not stop them from putting together such a great conference program.
Next stop is the CDKL5 Forum in November, also full of updates about the treatment programs in development for CDD. Hopefully by then we get to meet in person.
I hope you enjoyed this summary! let me know your thoughts in the comments.
Ana Mingorance, PhD
Disclaimer: These are my own impressions from the presentations that I was most interested in, and not an official text about the Alliance meeting by Alliance or the Loulou Foundation. I write these texts with the parents of people with CDD in mind, so excuse also my lack of technical accuracy in parts ;-)